
Abstract
In his report (Christison 1855) to the Royal Society of Edinburgh on 5th February 1855 of his pioneering studies upon the toxicology of the Calabar bean by self-experimentation, Robert Christison (see footnote 14 in Chap. 1) stated “that its active properties may be concentrated in an alcoholic extract, which constitutes 2.7 per-cent of the seed”. Although experimental details are not presented (Christison 1855), this deficiency was rectified some 8 years later by Thomas Fraser (see footnote 17 in Chap. 1), formerly Christison’s assistant at the University of Edinburgh (see footnote 14 in Chap. 1), who published the following method for the preparation of the tincture that he used in connection with his investigation into the therapeutic properties of the Calabar bean (Fraser TR 1863)
Notes
- 1.
Apothecaries or Troy ounce (Gunn and Carter 1965).
- 2.
Fluid ounces (Gunn and Carter 1965).
- 3.
A further manifestation of the widespread natural occurrence of P. venenosum throughout much of equatorial west and central Africa is apparent from a list (Neuwinger 1996) of the plant’s vernacular names which includes those in Cameroon, Central African Republic, Congo, Gabon, Ghana, Ivory Coast, Liberia, Sierra Leone and Zaire.
- 4.
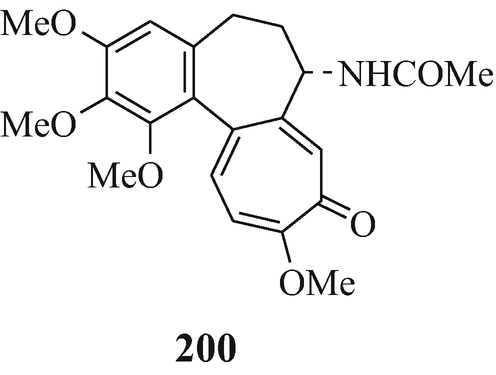
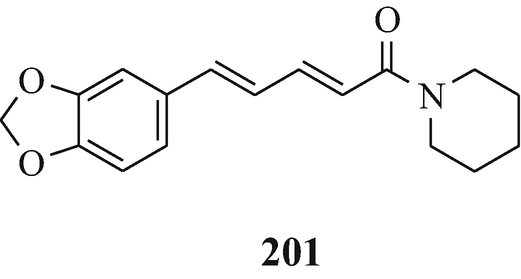
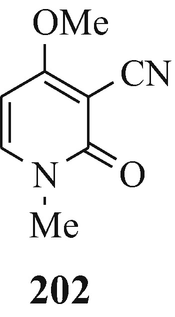
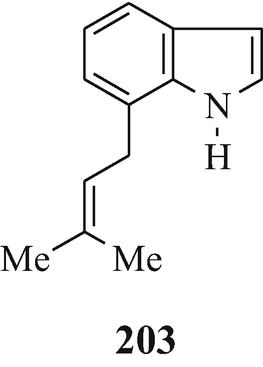
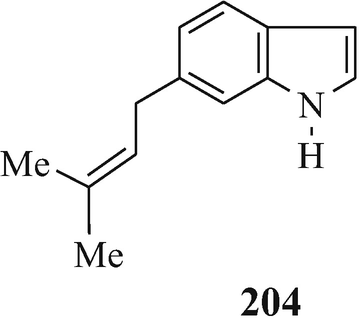
Toward the beginning of his most interesting and comprehensive deliberation upon “The Nature and Definition of an Alkaloid”, Pelletier (1983) notes that “The term alkaloid was coined in 1819 by the pharmacist W. Meissner and meant simply, alkalilike (Middle English alcaly, from Medieval Latin alcali, from Arabic alqaliy = ashes of saltwort, from qualey, to fry). The first modern definition by Winterstein and Trier described these substances in a broad sense as basic, nitrogen-containing compounds of either plant or animal origin” and for “True alkaloids” another four “additional qualifications” were defined. Pelletier later proceeds further by stating that “Therefore, following the concept that the structure of a compound [such as the presence of a basic nitrogen atom?] determines classification as an alkaloid, one should accept antibiotics of appropriate structure into the alkaloid class. Examples of antibiotics that, on the basis of structure, can be classed as alkaloids are .......... and the macrocyclic maytansinoids such as maytansine” but fails to note that although the structure of this latter compound contains three nitrogen atoms, like the two in the structurally closely-related maytanacine (1) they are all non-basic. He later elaborates upon this hypothesis by stating that “Though the term alkaloid originally meant alkalilike, we have seen that basicity can no longer be regarded as a necessary property of an alkaloid. Such glaring exceptions as colchicine [200], piperine [201], amine oxides......., and important quaternary salts such as laurifoline chloride......require that basicity no longer be included in a definition”. However, in this attempt to make the rule fit the exception rather than the exception fit the rule he meets opinions to the contrary from Wildman (1970) and Brossi (1997) who respectively state that “ Colchicine, C22H25NO6, is not an alkaloid in the strictest sense because the nitrogen atom is not basic being part of an acetamido function” [likewise, ricinine (202), isolated from the castor bean, Ricinus communis L. (Robinson 1988b), is not an alkaloid since one of its component nitrogen atoms is present in a non-basic lactam moiety – with the other being accounted for by a non-basic nitrile group] and “THE TERM “ALKALOID”, ORIGINALLY RESERVED FOR nitrogen-containing substances with basic properties found in plants, has a much broader meaning today and includes such substances occurring in mammals, fish, and mushrooms as well” (whilst, in the absence of opinion to the contrary, still retaining their basic properties), and also from the author who suggests a reappraisal of the alkaloidal status of the carbazole [so-called] alkaloids (Kapil 1971) since their component nitrogen atom is present in a non-basic carbazole moiety, as furthermore, is that in an essentially non-basic indole moiety [although the “weakly basic character of the indole nucleus” has been noted (Hinman and Lang 1960 – see also Robinson and Smith 1960) and was subsequently elaborated upon, in a review of the acidity [!] and basicity of indoles (Remers 1972), by the statement that “ the lone pair of electrons on the indole nitrogen is an integral part of the π-electron system and is not readily available for salt formation. A high concentration of hydrogen ions is therefore necessary to afford protonation of indoles. Such protonantion occurs mainly on C(3) in solution, but salts in which the proton was on nitrogen could be isolated from certain solutions by precipitation.”], of the two isomeric indole derivatives 203 and 204 which have been isolated from the liverwort, Riccardia sinuata (HOOK.) TREV (Benešová et al. 1969) and of other structurally similar naturally-occurring 6-isoprenylindoles (Ishii and Murakami 1975; Nwaji et al. 1972) which, contrary to their correct designation as natural products (Nwaji et al. 1972), have thus been erroneously referred to as alkaloids (Benešová et al. 1969; Ishii and Murakami 1975) – or even bases (Ishii 1981; Ishii and Murakami 1975). Despite the fact that a combination of both of the above structural features in 6-bromo-Nb-formyl-Nb-methyltryptamine would render both the nitrogen atoms non-basic, this component from the marine bryozoan Flustra foliacea (L.) has also been erroneously referred to as an alkaloid (Christophersen 1985b).
- 5.
Robert Robinson, OM, FRS (1885-1975)
The synthesis of brazilin [vide infra] would have no industrial value; its biological importance is problematical, but it is worth while to attempt it for the sufficient reason that we have no idea how to accomplish the task. There is a close analogy between organic chemistry in its relation to biochemistry and pure mathematics in its relation to physics. In both disciplines it is in the course of attack of the most difficult problems, without consideration of eventual applications, that new fundamental knowledge is most certainly garnered.
[Sir Robert Robinson (Farber 1963)]
Biographical (Farber 1963; Saltzman 1993; Sherwood Taylor 1948) and autobiographical (Robinson 1955, 1976) details relating to Robert Robinson have already appeared.
He was born on the 13th September 1885 in Bufford, near Chesterfield, Derbyshire, England, and it was in Chesterfield that his father, William Bradbury Robinson, was a highly successful manufacturer of cardboard boxes and surgical dressings and materials. Although his family were primary members of the Congregational Church in Chesterfield, Robert was a pupil at the Fulneck School, Pudsey Greenside, about midway between Bradford and Leeds, that was run by the Moravian Church in Great Britain – the majority of the masters were candidates for the Moravian ministry – and at which school he exhibited a natural predilection towards mathematics and physics and consequently wanted to be a mathematician. However, following his father’s wish, he decided to become a chemist and in 1902, upon passing the examination of the Joint Matriculation Board of the Universities of Manchester, Liverpool and Leeds, he became an undergraduate in the University of Manchester’s department of chemistry, then the paramount centre for both teaching and research in the subject throughout Great Britain. Although initially his chemical studies found him to be a somewhat reluctant student, it was during his second year at Manchester and consequent upon his attendance at lectures upon organic chemistry presented by William Henry Perkin jun, that the young Robinson became imbued with the subject and, after graduation in 1905 as Bachelor of Science with first-class honours in chemistry, he accepted a place in Perkin’s private laboratory. It would appear that it was from Perkin’s earlier research in the alkaloidal area that Robinson developed his interest in these natural products but, in the meantime, his work in his mentor’s laboratory led, in 1909, to his doctoral (DSc) thesis involving a study of the chemistry of brazilin (vide supra) and the related haematoxylin, the colouring matter of brazilwood. He was to prosecute this subject for the next six decades and, in fact, studies of the chemistry of brazilein were the first (Perkin jun and Robinson 1906) and last (Jaeger et al. 1974) [it is interesting that this paper was published from Shell Research Limited, Egham (vide infra)] of what were to be his some 700 contributions to the scientific literature between 1905 and 1974.
Robinson’s appointment to a junior position in the Department of Chemistry at Manchester in 1909 coincided with the acceptance of a senior position in that department by Arthur Lapworth who was to have a second major influence on Robinson’s career. His association with Lapworth between 1909 and 1912 led to Robinson’s interest in the theoretical aspects of organic chemistry to which he was no doubt predisposed in view of his early aptitudes to physics and mathematics. Ultimately, this association with Arthur Lapworth was to lead to Robinson’s development of a general theory of organic reaction mechanism, exemplified by the first use of the “curly arrow” to show electron movement (Kermack and Robinson 1922) and the introduction of the term “aromatic sextet” to denote the six-electron system that accounted for the unique properties of benzene (Armit and Robinson 1925).
In 1912 the Department of Chemistry at Manchester was to play a major personal role in Robert Robinson’s life when he married Gertrude Maud Walsh, one of the first research collaborators of one of his departmental colleagues, Dr Chaim Weizmann. Perhaps not surprisingly, this led to a life-long friendship between the Robinson and Weizmann families and, amongst other considerations, ultimately led – in December 1953, following the death of Weizmann at Rehovoth on 9th November 1952 and who had by then become the first president of Israel – to Sir Robert Robinson being invited to deliver the inaugural Weizmann Memorial Lectures which were ultimately to find publication as a unique and pioneering text (Robinson 1955) which still makes fascinating reading. It was during the period of the preparation of this presentation that Lady Gertrude Maud Robinson died suddenly on 1st March 1954 which elicited the following moving tribute from her husband – “She was the mainstay of my life and I owe to her encouragement and active help in every direction any little success that I have been able to achieve” (Robinson 1955). Indeed, early in what was to be a lifelong mutual collaboration, this Robinson duo was to publish (Robinson and Robinson 1918, 1924) (from the University of Liverpool and from the Universities of St Andrews and Manchester, respectively) (vide infra) what – unlike the postulations of others which were found to be inconsistent with subsequent experimental observations – has become, after further extension and interpretation in light of modern electronic theory, the currently accepted mechanism of that doyen of indole syntheses, namely that discovered by Emil Hermann (Lucier 1993) Fischer (Robinson 1982). The three-stage mechanism which they proposed essentially embodies an earlier concept by Brunner (1898b) in which he “had made a comparison between the acid catalysed conversion of 2,2'-diaminobiphenyl into carbazole and the acid catalysed indolization of methyl isopropyl ketone Nα-methylphenylhdrazone” (Robinson 1982) ˗ but which was not acknowledged by the Robinson duo. In addition to giving chemical analogies for all the three stages of their mechanistic proposal, the Robinsons, furthermore, extended their theory to the synthesis of 2,3,4,5-tetraphenylpyrrole from bisbenzyl phenyl ketone azine (Robinson and Robinson 1918). However, unfortunately, as they appear to have been with Brunner’s 1898 paper, they were also apparently unaware of another German publication (Piloty 1910) which had reported the acid-catalysed conversion of bisdiethyl ketone azine into 2,5-diethyl-3,4-dimethylpyrrole. It is obvious from their publications, that the Robinsons had access to the German literature covering these two German publications and their apparent unawareness of them is, therefore, surprising (Robinson 1982) – for the second enigma in Robert Robinson’s scientific studies, see Sect. 3.1 and vide infra.
Also in 1912, Robert Robinson was offered, and accepted, his first professorship – at the University of Sydney – but in 1916 he returned to England to the newly-created Heath Harrison Chair of Organic Chemistry at the University of Liverpool. Here, he had the opportunity to renew his collaboration with Perkin, working on various aspects of alkaloid chemistry [see, for example (Perkin jun and Robinson 1919) from the Universities of Oxford and Liverpool], which led to his first success in this area by the synthesis of tropinone (which is structurally-related to atropine and cocaine) by reaction between acetone, methylamine and succindialdehyde (Robinson 1917a). This was published from the University of Liverpool, as was a later paper (Robinson 1917b) which concerned his first speculations relating to alkaloid biogenesis. Whilst in Liverpool, Robert Robinson also participated in research directly related to the World War I (the so called “war to end all wars”) effort of rejuvenating the dyestuffs industry and this led, in 1919, to his full-time appointment as Director of Research at British Dyestuffs in Huddersfield. However, owing to the problems in management he soon resigned from this post when the Chair of Chemistry became available at the University of St Andrews in 1920 – in which year he was also elected a Fellow of the Royal Society. Whilst at St Andrews he was able to return to the study of alkaloids and natural colouring matters and also began to seriously formulate his ideas relating to an electronic theory of mechanism of organic reactions based upon the ideas of those including Arthur Lapworth (vide supra). However, his stay at St Andrews was curtailed in 1922 when he was offered, and accepted, the Chair or Organic Chemistry at Manchester subsequent to Lapworth being appointed as Head of the Department. During this second period at Manchester of some six years, there was fully developed an electronic theory of organic chemistry and, furthermore, many studies consolidating previous investigations on various alkaloids appeared. Included in these were the inaugural papers (Robinson and Suginome 1932a, b) relating to a potential synthesis of l-physostigmine (Sect. 3.1). The scene was now set for Robinson’s final moves – in 1928 to University College, London – from where the results of studies involved in the verification of the structure of l-physostigmine were published (Boyd-Barrett and Robinson 1932) (see footnote 10 – as quoted in Sect. 2) – and ultimately, in 1930, upon the death of Perkin in 1929, to the Waynflete Professorship of Chemistry at the University of Oxford (Magdalen College) where, amongst many other investigations, were to be the continuation of the quest for synthetic l-physostigmine which, although ultimately without success (Sect. 3.1), was to present an enigma that has only recently (Robinson 2002 – see also Ault 2008 and Sect. 3.1) been clarified.
Robinson’s first decade at Oxford also saw the completion of work begun earlier on the plant pigments anthoxanins and anthocyanins and the beginning of work upon synthetic approaches to steroids and in 1939 he received a knighthood and was elected as President of the Chemical Society. The outbreak of World War II witnessed the beginning of his work – in association with Howard W Florey and Ernst Chain – on the chemistry of penicillin which continued until the declaration of peace at which juncture he returned to his research on the synthesis of steroids and, being mindful of the results from degradative work already effected by Hermann Leuchs, turned his attention to the establishment of the structures of strychnine and brucine, success in which was one of his major contributions to the study of the alkaloids. Indeed, in the main publication (Woodward et al. 1963) presenting the verification of the structure of the former alkaloid by total synthesis, it is stated with regard to its structural elucidation that “over a period of forty years, one of the great classics of structural organic chemistry was constructed. In that effort, described in more than two hundred and fifty separate communications, Robert Robinson played a brilliant and commanding role, and the extensive beautiful experimental contributions of Hermann Leuchs were of definitive importance.” In 1947 Robert Robinson was awarded the Nobel Prize in Chemistry (Farber 1963; Saltzman 1993) and elected President of the Royal Society. Perhaps it is not surprising that he became one of the major spokespersons for science and in 1949 he received that most prestigious civilian decoration in the UK when he was elected to the Order of Merit.
His tenure was extended at the University of Oxford until 1955 but at this juncture he became a director of Shell Chemical Co Ltd when a small laboratory was placed at his disposal and he took a residence at 170 Piccadilly, London W1. As a consultant he visited the research establishments of Shell and continued in this role until he died in 1975.
- 6.
Prior to the advent of 1H-nmr spectroscopy, the quantitative estimation of methoxy and methylimino groups was based upon a technique that involved initially heating with concentrated hydriodic acid. Under these conditions at 100°C, cleavage of methyl ether groups occurred to afford iodomethane (leading to the Zeisel method) (Zeisel 1895), as does likewise that of methylimino groups when the temperature was raised to 150 °C (leading to the Herzig-Meyer method) (Herzig and Meyer 1897). Thus, methoxy and methylimino groups could be estimated separately when both were present in the same compound and when, in both cases, the liberated iodomethane was sequentially driven off with carbon dioxide, absorbed in an ethanolic solution of silver nitrate and the silver iodide so formed determined gravimetrically. Alternatively, the iodomethane was pyrolysed to afford iodine which was determined volumetrically by sequential dissolution in potassium iodide solution and titration against thiosulphate. Both these approaches have been the subject of thorough reviews (Anon 1945; Kingscott and Knight 1914; Niederl and Niederl 1948).
“Great caution has”, however “to be exercised in interpreting the results. Some substances yielded only one-half or one-third of their number of N-alkyl radicals, such as certain alkaloids and complex N-heterocyclic compounds” (Niederl and Niederl 1948 ˗ see also Anon 1945). Thus, using this technique with l-physostigmine initially indicated the presence of only two of the N-methyl groups and it was only when the product remaining after the removal of those two methyl groups was examined further by the technique that the presence of the third N-methyl group became apparent (Herzig and Lieb 1918a, b). Similary, only one of the two N-methyl groups in eseroline (3, R1=H, R2=Me, X=NMe) was detected (Salway 1912b) by application of the Herzig-Meyer (1897) method, the second only becoming apparent (Straus 1914) by use of the Pregl apparatus – which is probably that illustrated in (Anon 1945). It appears that the accurate application of this technique depends upon the proportion of hydriodic acid used (Herzig and Lieb 1918a, b) and its quality (Niederl and Niederl 1948).
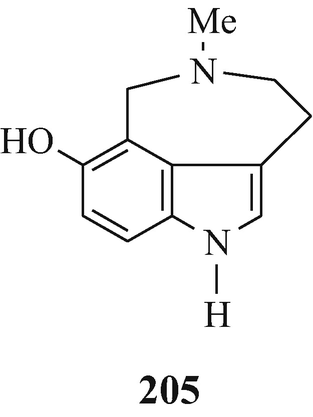
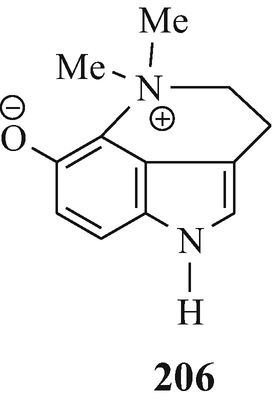
This may also account for the detection (Jensen and Chen 1936) of only one N-methyl group (unfortunately experimental details were not presented) in dehydrobufotenine {the principle indolic component isolable from the parotid glands of the South American toad Bufo marinus (Märki et al. 1961) and one of the components (as its O-sulphate, bufothionine) [Spande 1979(h)] of the skin of Bufo marinus (Honda et al. 1991) and of Bufo bufo bufo (Robinson et al. 1961) and also found in other Bufo sources [Spande 1979(h)] and in a galenical preparation of one of these known in China as Ch’an Su and in Japan as Senso [Robinson 1917b, 1960 ˗ see, however, Jensen and Chen 1936; Spande 1979(h)]} that led to the erroneous postulation of 205 as its structure (Robinson 1917b, 1960). This, from earlier chemical and uv spectroscopic properties (Witkop 1956) and primarily a detailed study of its 1H-nmr spectrum, was subsequently corrected to 206 [Märki et al. 1961, Robinson et al. 1961 – see also Spande 1979(c)] which was ultimately confirmed by the synthesis of dehydrobufotenine (Gannon et al. 1967) and later by X-ray crystallography of bufothionine (Honda et al. 1991).
- 7.
A facile conversion of l-physostigmine into l-eseroline (obtained as its fumarate salt in 98% yield and from which the free base can readily be isolated) in refluxing butan-1-ol in the presence of a catalytic amount of sodium butoxide was later reported (Brossi 1990; Brossi and Yu 1988b – see also 1988a; Yu et al. 1987, 1988b). This was particularly apposite since l-eseroline “ is extremely sensitive to aerial oxidation” and its preparation by either alkaline or acid hydrolysis of l-physostigmine requires “ neutralization and work-up from aqueous solutions, and proved tedious when carried out on a larger scale” (Yu et al. 1987).
- 8.
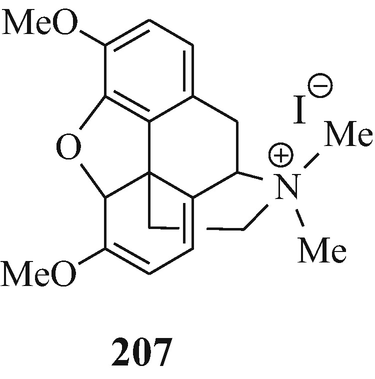
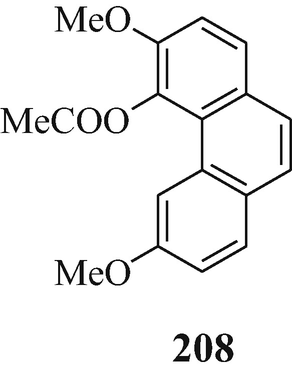
The decomposition of eserethole methiodide to yield physostigmol ethyl ether is now known to be analogous to the reaction of thebaine methiodide (207) when heated in the presence of acetic anhydride to afford the product 208 – both cases involve the loss of the fragment CH2CH2⨁ NMe2IƟ and result in the formation of an aromatic system (Coxworth 1965).
- 9.
This was the first recognition of the natural occurrence of the Ph-N-C-N system (Robinson 1963a) and the 1,2,3,3a,8,8a–hexahydropyrrolo[2,3-b]indole ring system which have since been detected in other products which have been isolated and characterised from plants, fungi and animals. Such metabolites comprise:-
-
9.1.
Two of the Calabar bean’s other structurally closely-related alkaloids, namely l-eseramine (98, R1=MeNHCO, R2=Me, X=NCONHMe, n=1) and l-N(8)–norphysostigmine (98, R1=MeNHCO, R2=H, X=NMe, n=1) (Chaps. 4 and 5 respectively.
-
9.2.
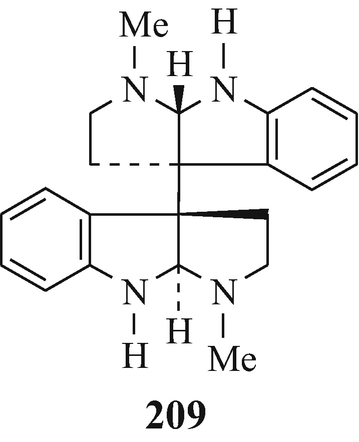
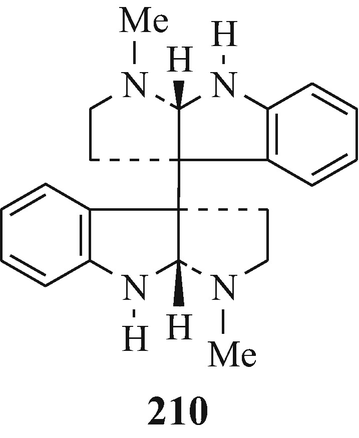
The dimeric tryptamine-derived alkaloids chimonanthine (122, R1=R2=H) (Grant et al. 1962, 1965; Hendrickson et al. 1962; Hino and Nakagawa 1988; Hodson et al. 1961; Kutney 1977; Robinson 1963a; Scott et al. 1964; Taylor 1966), calicanthidine (122, R1=H, R2=Me) (Grant et al. 1962, 1965; Hino and Nakagawa 1988; Saxton et al. 1962; Robinson 1963a; Scott et al. 1964; Taylor 1966) and folicanthine (122, R1=R2=Me) (Grant et al. 1962, 1965; Hino 1961a, b; Hino and Nakagawa 1988; Hino and Yamada 1963; Hodson and Smith 1956; 1957; Kutney 1977; Robinson 1963a; Taylor 1966) ˗ all three of which have been isolated from plants in the botanical order of Calycanthaceae, a small family comprised only of the genera Calycanthus and Chimonanthus (Cordell 1981) [chimonanthine (122, R1=R2=H) has also been isolated from Palicourea fendleri (Hino and Nakagawa 1988; Nakano and Martin 1976) and P domingensis (Hino and Nawagawa 1988; Ripperger 1982), meso-chimonanthine (209) has been found along with chimonanthine (Gorman et al. 1971; Hall et al. 1967) in Chimonanthus fragrans, Lindle ((Meratia praecox, Rehd and Wils.), the deciduous shrub commonly known as Winter Sweet (Grant et al. 1965), and the enantiomer of chimonanthine, namely d-chimonanthine (210), has been isolated from the skin of Phyllobates terribilis (the Columbian poison-dart frog) (Hino and Nakagawa 1988; Tokuyama and Daly 1983).
-
9.3.
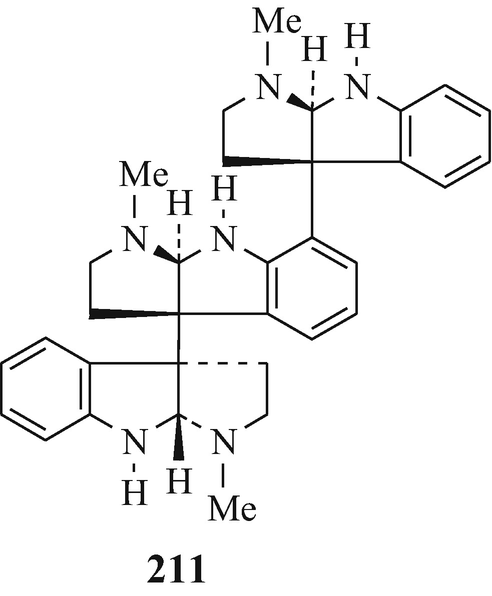
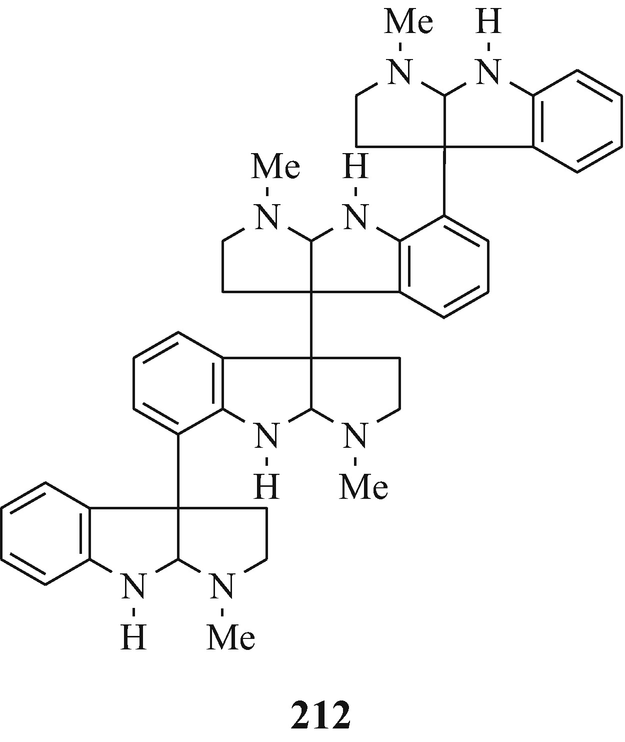
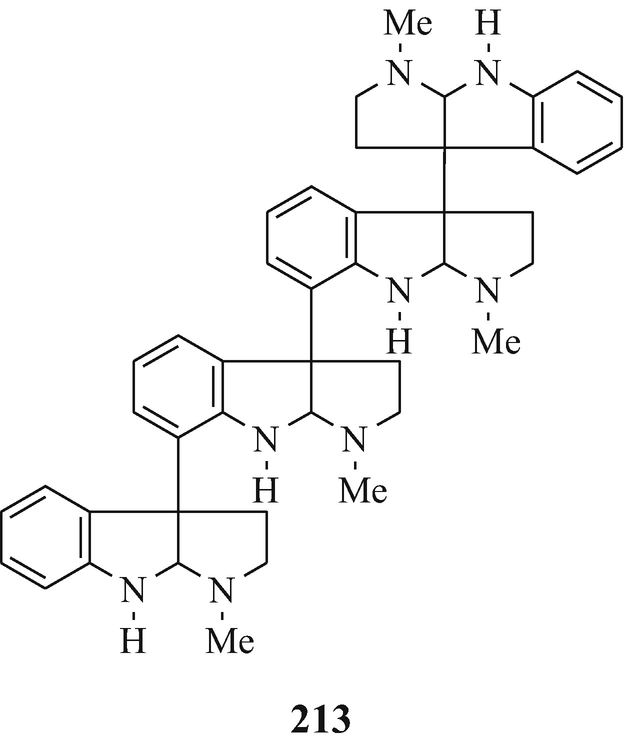
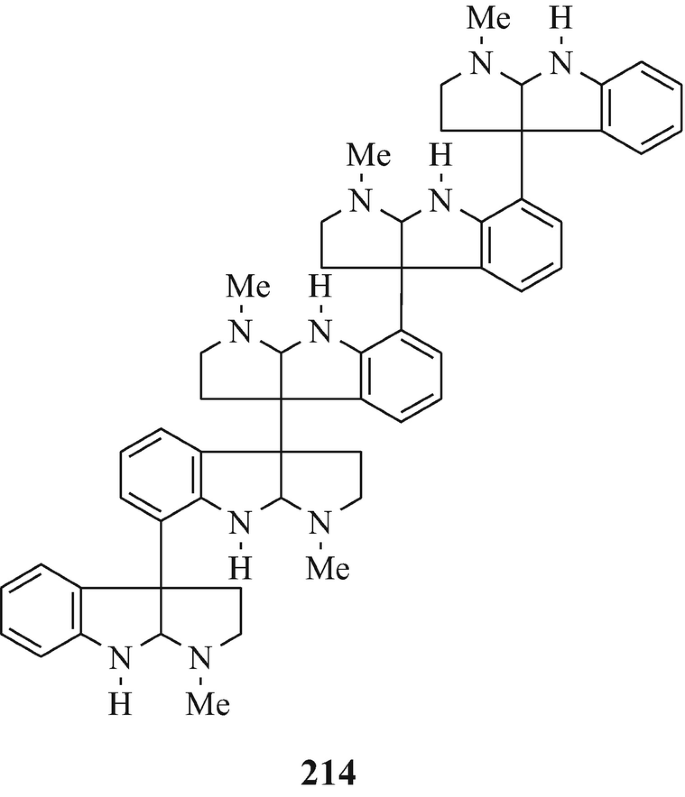
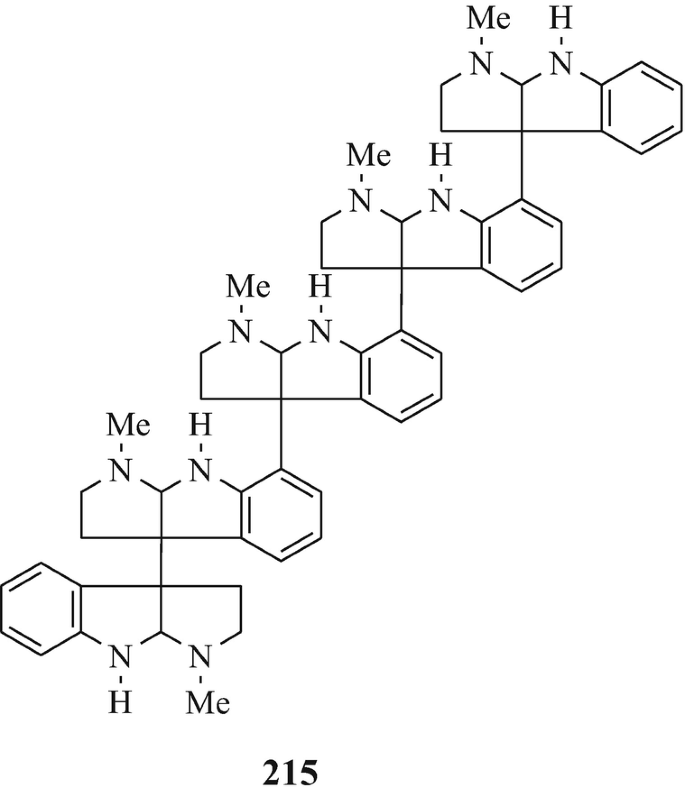
The next higher oligomers, namely the trimer hodgkinsine (211) (Attitulah et al. 1966; Fridrichsons et al. 1967, 1974; Gorman et al. 1971; Hino and Nakagawa 1988) and the tetramers quadrigemine-A (212) (as a mixture of diastereoisomers) and quadrigemine-B (213) (Gorman et al. 1971; Hino and Nakagawa 1988; Parry and Smith 1978) [all three of which have been isolated from Hodgkinsonia frutescens F.Muell, “ a plant in the family Rubiaceae quite unrelated to the Calycanthaceae” (Cordell 1981)], and the alkaloids with the highest molecular weight so far in this series, namely the pentamers psychotridine (214) which was isolated from Psychotria beccarioides Wernh, also of the family Rubiaceae (Hart et al. 1974; Hino and Nakagawa 1988) and isopsychotridine – tentatively assigned structure (215) and isolated from Psychotria forsteriana A.Gray along with psychotridine (214) and quadrigemines-A and -B (212 and 213, respectively) (Hesse 1981; Hino and Nakagawa 1988; Roth et al. 1985). It is interesting that the statement made consequent upon the elaboration of the structure of hodgkinsine (211) (Fridrichsons et al. 1974) that “the possibilities appear to exist for molecules of greater polymeric elaboration involving 4, 5…… N-methyltryptamine units” (Fridrichsons et al. 1974) thereby predicted the natural occurrence of structures such as 212, 213, 214, and 215. Perhaps higher oligomers, and even polymers – possibly having structural functions ˗ remain to be discovered! However, the parent monomeric tricyclic moiety has been somewhat elusive (see footnote 4 in Chap. 3).
-
9.4.
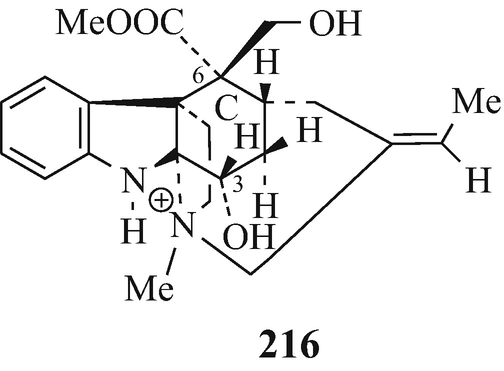
Some of the monoterpenoid indole alkaloids such as borreverine [Cordell 1983(a), Herbert 1983(c)], cabuamine [Joule 1983(e)], corymine [Joule 1983(cf), Robinson 1963a], echitamine salt [Creasey 1983(c); Herbert 1983(a); Joule 1983(d)] [in this category, this was the first of these alkaloids – although as a quaternary salt, and in the strictest sense, it is therefore not an alkaloid (see footnote 4) – of which the structural elucidation (as 216, “the disposition of the groups being with respect to the substituted cyclohexane ring C, which is in the boat form with C3 and C6 above the plane of the paper”) was effected – this work has been comprehensively reviewed (Robinson 1963a; see also Hamilton et al. 1962)], erinicine [Joule 1983(g)], erinine [Joule 1983(fg)], eripine [Joule 1983(c)], hunteracine [Herbert 1983(b), Husson 1983], isocorymine [Joule(c)], peceylanine, peceyline [Cordell 1983(c)], pelankine [Cordell 1983(d)], pleiocorine [Cordell 1983 (b)], rhazidine salt (Hino and Nakagawa 1988; Saxton 1983b) and vincorine [Cordell 1983(b), Joule 1983(e)].
-
9.5.
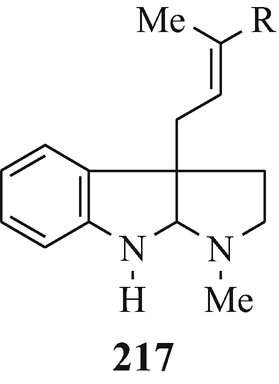
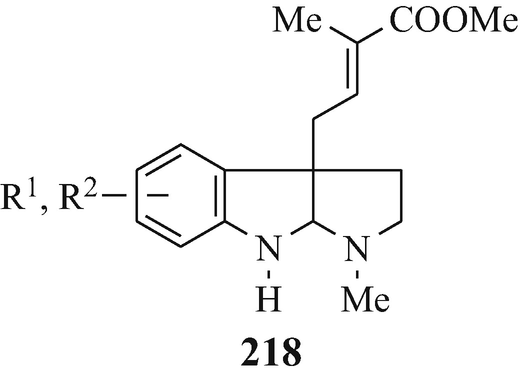
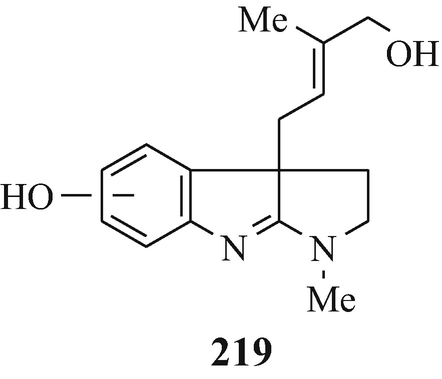
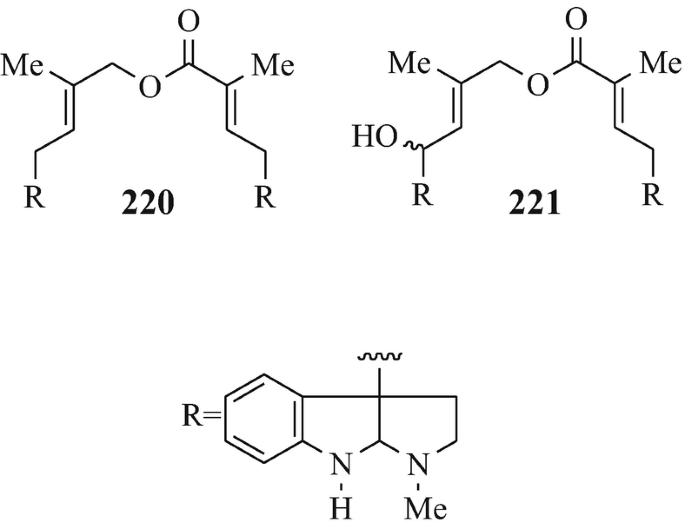
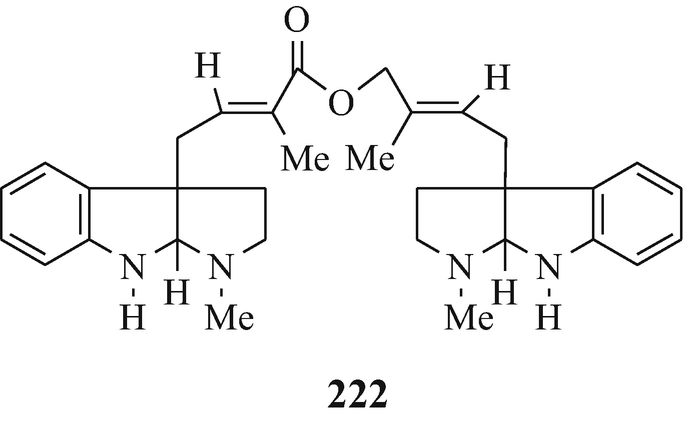
Isolated from Australian frogs of the genus Pseudophryne (of the family Myobatrachidae), a group of pseudophrynamines, namely 217 [R=CH2OH, COOMe and CHO (tentative structure)] (Daly et al. 1990), 218 (R1=H, R2=OH; R1=H, R2=MeO; R1=OH, R2=MeO and R1=R2=MeO) (Daly et al. 1990), 219 (Daly et al. 1990), 142 [R=H and Me (both tentative structures)] (Daly et al. 1990), 220 (Daly et al. 1990) and 221 (tentative structure) (Daly et al. 1990) and, found in the skin of the Australian frog Pseudophryne coriacea, 217 (R=CH2OH) (Crich et al. 1995; Mitchell and Le Quesne 1990; Spande et al. 1988), 217 (R=COOMe) (Spande et al. 1988) and 222 (≡220) (Cozzi et al. 1990; Mitchell and Le Quesne 1990; Spande et al. 1988).
-
9.6.
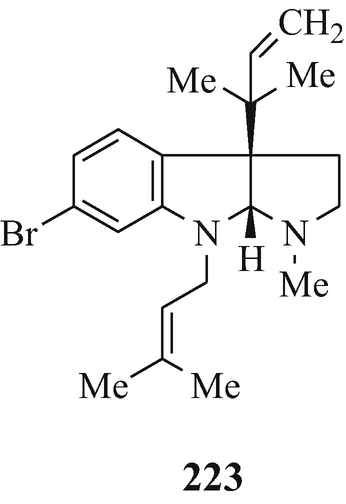
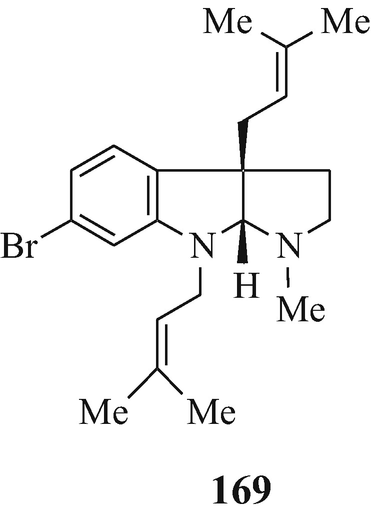
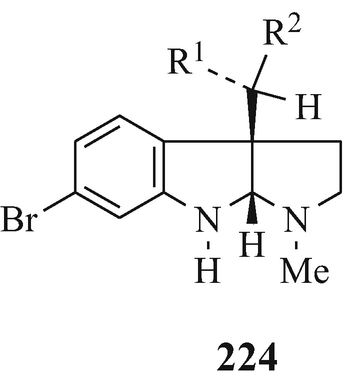
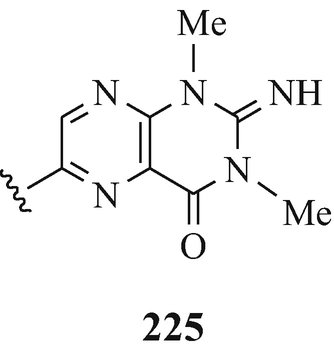
A series of 6-bromo-1,2,3,3a,8,8a-hexahydroypyrrolo[2,3-b]indoles which are also usually 3a-prenylated [for example, flustramine A (223) (Carlé and Christophersen 1979, 1980) and flustramine B (169) (Carlé and Christophersen 1979, 1980; Muthusubramanian et al. 1983; Hino et al. 1983] and which have been isolated from the marine bryozoan Flustra foliacea (L) and have already been the subject of reviews (Christophersen 1983, 1985a, b, Hino and Nakagawa 1988) and two of which, urochordamine A and B (224, R1=Et, R2=225) and (224, R1=225, R2=Et), respectively, have been isolated from the tunic (outer body) of the adult tunicate Ciona savignyi (a marine animal) and which promote settlement and metamorphosis of the C. savignyi larvae (Tsukamoto et al. 1993).
-
9.7.
From a variety of sources, microbial metabolites (several of which have biological activity which suggests a potential either therapeutic or other utility) which include:-
-
9.7.1.
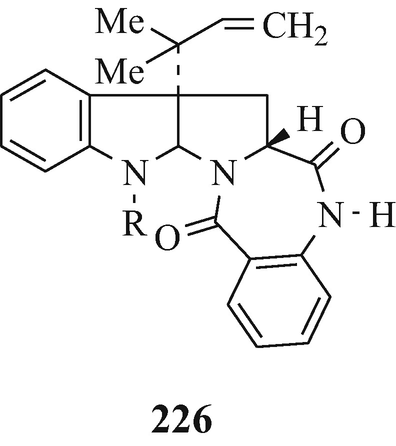
Aszonalenin (226, R=H) which “apparently induced the abnormal second cleavage the sea urchin embryos” and which was isolated, along with LL S490β (226, R=COMe), from Aspergillus zonatus IFO 8817 (Hino and Nakagawa 1988; Kimura et al. 1982), with the latter metabolite having earlier (Ellestad et al. 1973) been isolated from an unidentified Aspergillus species.
-
9.7.2.
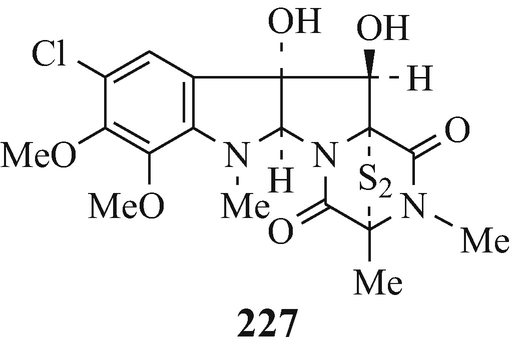
Responsible for the serious animal disease of “facial eczema” (Hino and Nakagawa 1988), which in sheep causes extensive liver damage and ultimate death and which is widespread in New Zealand with outbreaks having also occurred in Australia, sporidesmin A (227) (Fridrichsons and Mathieson 1965), along with the structurally closely-related sporidesmins B, C, D, E, F and G, has been isolated from the fungus Pithomyces chartarum (Hino and Nakagawa 1988; Sammes 1975).
-
9.7.3.
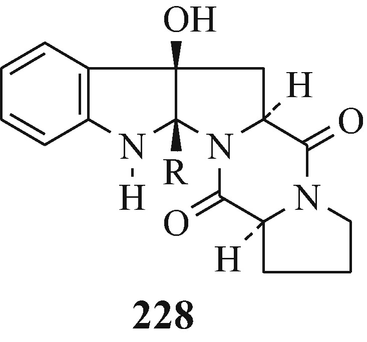
Brevianamide E (228, R=CMe2CH=CH2) (Hino and Nakagawa 1988; Sammes 1975) which was isolated from the mould Penicillum brevi-compactum Dierckx (University of Manchester Acc 382) (Birch and Russell 1972; Birch and Wright 1970; Christophersen 1983).
-
9.7.4.
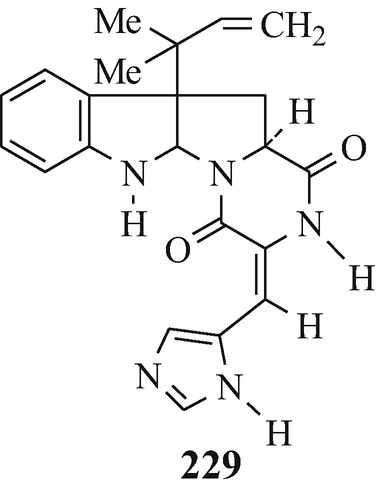
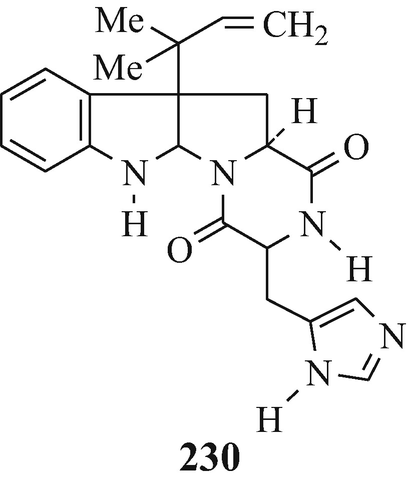
Roquefortine (229), a neurotoxic mycotoxin which showed tremorgenic activity in male mice [LD50 (i.p.) 15-20mg/kg, with doses of 50-100mg/kg causing prostration and an atonic posture (Scott et al. 1976)], was isolated from Penicillium roqueforti (Christophersen 1983; Hino and Nakagawa 1988; Scott et al. 1976; Vleggaar and Wessels 1980) –it was also reported (Ohmomo et al. 1975, 1977, 1978) as being isolated (as roquefortine C) from this same source and as a metabolite of P crustosum (Hino and Nakagawa 1988; de Jesus et al. 1983). Reduction of roquefortine C using zinc powder in boiling acetic acid under reflux afforded a mixture of two stereoisomers of dihydroroquefortine C (230) which were separated by column chromatography on silica gel. Although the absolute configuration at the newly-formed asymmetric carbon atom remains to be established in both these isomeric products, one of them was found to be identical with the roquefortine D which was also isolated from P roquefortii – interestingly, the other isomer could not be found in this source (Ohmomo et al. 1978).
-
9.7.5.
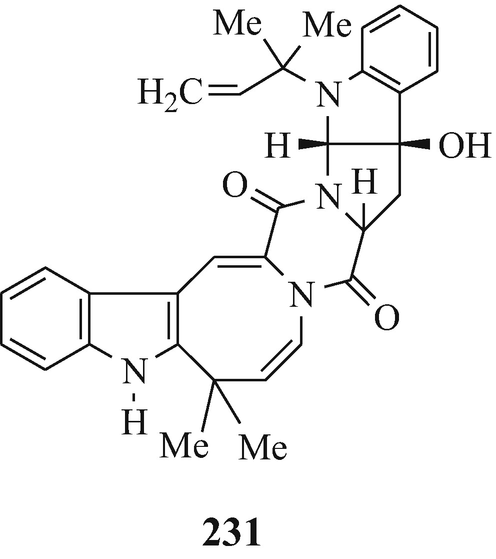
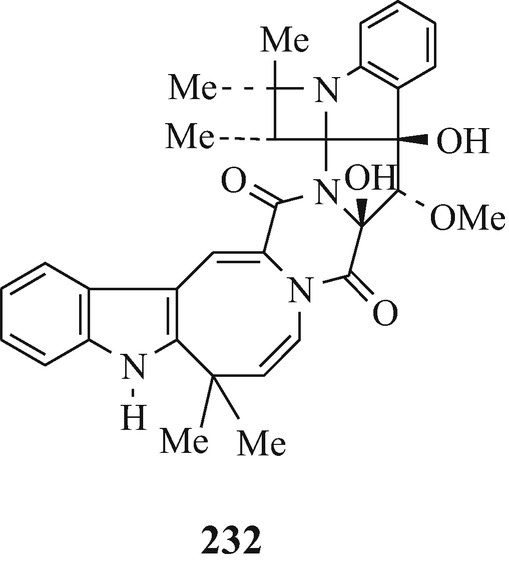
Okaramine A (231) and okaramine B (232), produced by penicillium simplicissium AK-40 (isolated from a soil sample) when cultured with okara (the insoluble residue of whole soybean) (Hayashi et al. 1989). Both the metabolites exhibited insecticidal activity against the 3rd instar larvae of silkworm, with okaramine B being much more active than okaramine A which was as active as physostigmine (Sect. 10.8) (Hayashi et al. 1989).
-
9.7.6.
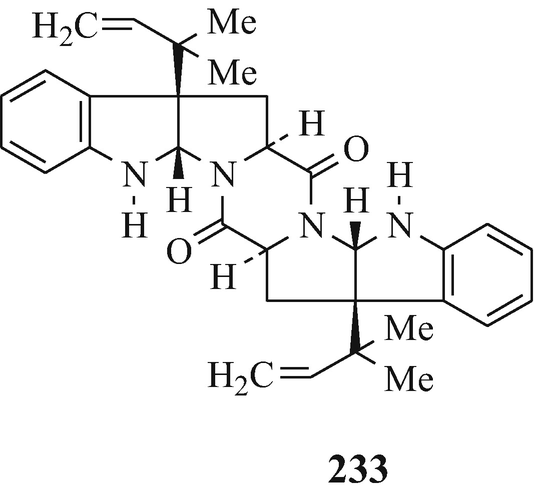
With vasodilating activity, although further details of its pharmacological characterisation await publication, amauromine was isolated from the culture broth of Amauroascus sp. No.6237 (Takase et al. 1984) and its structure was determined as (233) from chemical and spectral data (Takase et al. 1985b) and verified as such by total synthesis (Takase et al. 1985a, 1986a, b). It “ is the first natural diketopiperazine composed of two L-tryptophans” (Hino and Nakagawa 1988) and also appears to have been reported as a metabolite of Penicilliun nigricans, but with no stereochemistry indicated, under the name of nigrifortine which was recognised as “the first example of a prenylated diketopiperazine fungal metabolite in the form of an indolic symmetrical dimer” (Laws and Mantle 1985).
-
9.7.7.
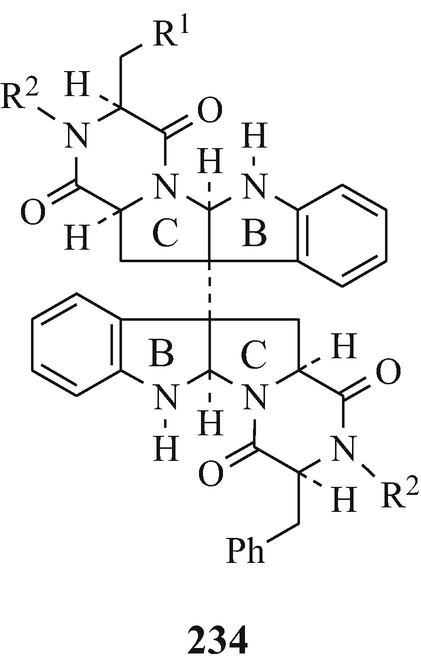
Two dimers WIN 64821 (234, R1=Ph, R2=H) and WIN 64745 (234, R1=CHMe2, R2=H), obtained from the whole culture fermentation broths of Aspergillus sp,SC319 (ATCC 74177) originally isolated from soil, with the former metabolite being found to be “ a competitive antagonist to substance P(SP) at the human NK1 receptor with an inhibitor affinity constant (Ki) of 230± 30nM against [125I] SP in human astrocytoma cells” (Barrow et al. 1993).
-
9.7.8.
A group of structurally-related fungal metabolites which together have been subjected to review (Hino and Nakagawa 1988) and include:-
-
9.7.8.1.
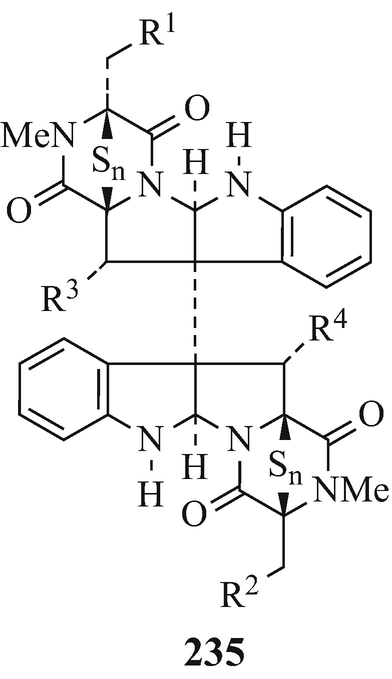
Chaetocin (235, R1=R2 = OH, R3=R4=H, n=2) (see also Sammes 1975; Weber 1972) isolated from Chaetomium minutum and possesses antibacterial and cytostatic activity.
-
9.7.8.2.
The antibiotics verticillines A (235, R1=R2=H, R3=R4=OH, n=2) and B (235, R1=H, R2-R4=OH, n=2) which were isolated from Verticillium sp.(strain TM-759) and possess antimicrobial activity against gram-positive bacteria and mycobacteria and are cytotoxic, both with ED50 against HeLa cells of 0.2μ/ml.
-
9.7.8.3.
Produced by Acrostalagmus cinnabarinus var. melinacidinus, melinacidins II (235, R1=R3=H, R2=R4=OH, n=2), III (235, R1-R3=OH, R4=H, n=2) and IV (235, R1-R4=OH, n=2) which inhibit the growth of a variety of gram-positive bacteria in vitro. A similar metabolite, chetracin A (235, R1-R4=OH, n=4) which “exhibits remarkable cytotoxicity to HeLa cells” has been isolated from Chaetomium abuense Lodha and C. retardatum Carter et Khan.
-
9.7.8.4.
Obtained from Aspergillus flavus (strain MIT-M-25, 26 and 27) and A. Flavus var columnaris, ditryptophenaline (234, R1=Ph R2=Me, with optical asymmetry at the B/C ring junction inverted) which possesses neither significantly toxic (LD50 200mg/kg) nor antibiotic properties.
-
9.7.8.1.
-
9.7.9.
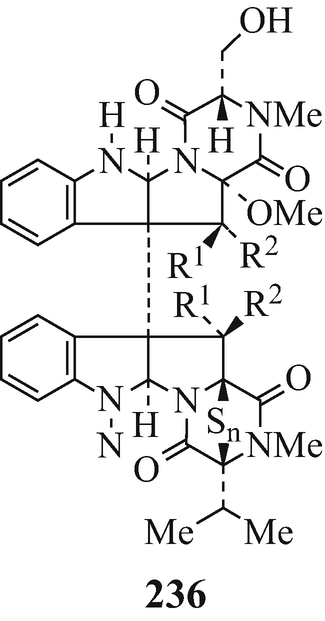
Isolated from a strain of Leptosphaeris sp. originally separated from the marine alga Sargassum tortile, leptosins M and M1 (236, R1=OH, R2=H, n=4 and 2, respectively) and N and N1 (236, R1=H, R2=OH, n=4 and 3, respectively). Significant cytotoxicity against the murine P388 cell line was exhibited by all four of these metabolites (with the activities of leptosins N and N1 being almost 10x greater than those of leptosins M and M1), as it was against a disease-oriented panel of 39 human cancer cell lines. Interestingly, it appears from further investigations that the modus operandi of leptosin M is probably different from that of any other currently known oncolytic drugs (Yamada et al. 2002).
-
9.7.10.
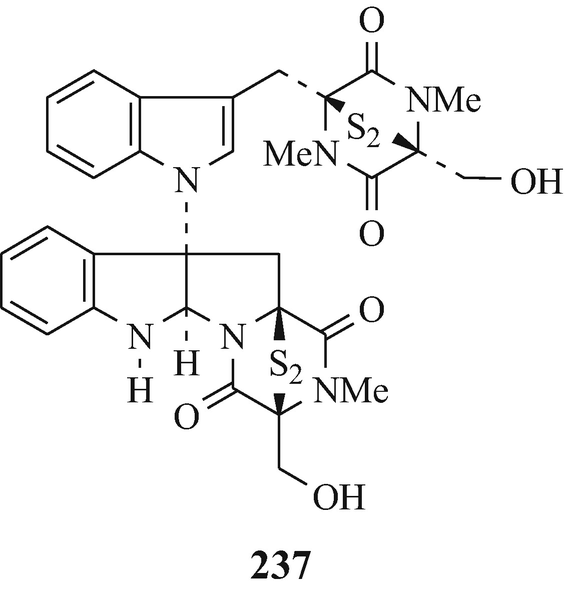
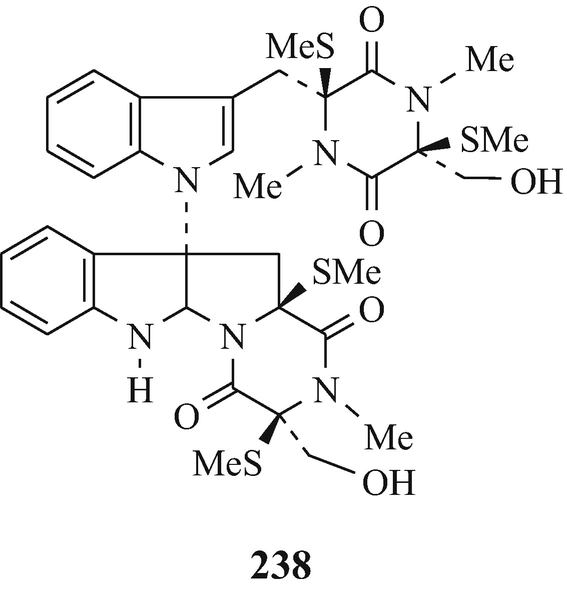
Chetomin (237), a toxic metabolite which is strongly cytotoxic towards HeLa cells, is produced by various Chaetomium species, including C. abuens, C. cochliodes, C. globosum, C. retardum and C. tenuissimum, and dethio-tetramethylthiochetomin (238) has been isolated from C. globosum Kinze ex Fries (Hino and Nakagawa 1988, Kikuchi et al. 1982).
-
9.7.1.
-
9.1.
- 10.
This pioneering synthesis from University College, London (see footnote 5) (Boyd-Barrett and Robinson 1932) began with the Fischer indolisation in warm alcoholic sulphuric acid of the phenylhydrazone 12 (R=H) that was formed in situ – from phenylhydrazine and 2-oxo-5-phenoxypentane – to afford 13 (R=H) { the possible alterative structure 14 (R=H) was eliminated since “ the colour reactions [with Erhlich’s reagent] do not indicate the presence of a free position in the pyrrolo nucleus” and “a further argument is that, in the later stages,” 14 (R=H) “would lead to products with an additional methylene group, and this is not indicated by the analyses”}. Compound 13 (R=H) was then heated in an autoclave with iodomethane in methanol at 120oC for 8h to yield 15 (R=H) which upon shaking with a mixture of aqueous sodium hydroxide and ether for 7h gave 11 (R1=H, R2+R3=CH2, R4=OPh) that in cooled acetone was oxidised by the gradual addition of finely powdered potassium permanganate over 9h to afford 11 ((R1=H, R2+R3=O, R4=OPh) (see footnote 11). Heating this under reflux with hydrobromic acid (d 1.7) for 12h yielded 11 (R1=H, R2+R3=O, R4=Br) which upon heating in methanol in a sealed tube at 170-180oC for 10h with aqueous dimethylamine afforded 11 (R1=H, R2+R3=O, R4=NMe2) and likewise with aqueous methylamine afforded 11 (R1=H, R2+R3=O, R4=NH Me).
This latter product was also a key intermediate in the Julian and Pikl synthetic approach to the physostigmime ring system (Sect. 3.1) and the above studies therefore represent an alternative route to its synthesis, albeit “a very complicated route” as stated by the American duo (Julian and Pikl 1935a).
Moreover, and concomitantly with the completion of this synthesis and with reference to the latter product it was stated that “We are of the opinion that it should be possible to effect the ring closure of substances of type (VIII) [11 (R1=H, R2+R3=O, R4=NHMe)], but this has not yet been investigated” (Boyd-Barrett and Robinson 1932) ˗ and, it has been claimed (Brossi et al. 1996) “was never completed.” However, later in that same year such an investigation was, in fact, reported by King and Robinson (1932b) although they incorrectly identified the product from their reaction and thereby failed to recognise the success of their efforts (Sect. 3.1)
It may, however, be significant that the above opinion by Boyd-Barrett and Robert Robinson (1932) [and a similar one expressed by King and Robert Robinson (1932b)] would have been available to Julian and Pikl either before they embarked upon their synthetic studies toward l- physostigmine or during its early phases, in which this type of ring closure is fundamental. Indeed, the paper by Boyd-Barrett and Robert Robinson (1932) is quoted as a reference by Julian and Pikl (1935a). Consequently, it is possible that Percy Julian could have deliberately engineered the altercation (see footnote 12) that was to become “the very public disagreement between himself and Robert Robinson” (Ault 2008).
- 11.
The colourless 1,3,3-trimethyl-2- methyleneindoline (250, R1+R2=CH2) upon exposure to air and sunlight rapidly began to redden [as had been observed previously (Brunner 1898a, b, Plancher 1898) with this compound] and was, over a period of 10 days, completely autoxidised into 250 (R1+R2=O) and formaldehyde (Robinson 1962, 1963b). By analogy with these observations, it is now suggested that the enamine 11 (R1=MeO, R2+R3=CH2, R4=OPh) which, as also reported (King and Robinson 1932a), “acquired a purple tint in the air” [see also (Boyd-Barrett and Robinson 1932) for an analogous observation with 11 (R1=H, R2+R3=CH2, R4=OPh) and (Boyd-Barrett 1932; Ficken and Kendall 1961; Robinson 1960) for other examples of related enamines which have also been observed to redden upon aerial exposure and thereby likely undergo similar autoxidation], is likely to be similarily converted into 11 (R1=MeO, R2+R3=O, R4=OPh). This would thereby obviate the use of potassium permanganate to effect this transformation (King and Robinson 1932a) and that of 11 (R1=H, R2+R3=CH2, R4=OPh) into 11 (R1=H, R2+R3=O, R4=OPh) (Boyd-Barrett and Robinson 1932).
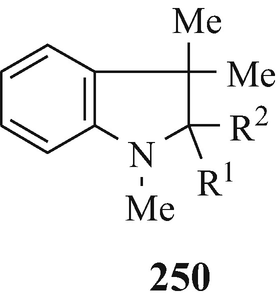
It has not escaped attention that similar autoxidations of either such enamines or of tertiary indolinols {exemplified by the simple analogue 2-t-butyl-1,3,3-trimethylindolin-2-ol 250 (R1=tBu, R2=OH) which also undergoes an “interesting” [Spande 1979(e)] air-oxidation to afford 250 (R1+R2=O) with loss of the t-butyl substitutuent – indolinols with 2-phenyl and 2-hydrogen substitutents are stable (Robinson 1962, 1963b)} could be involved in the formation (including biosynthetic) of alkaloids containing the indolin-2-one moiety, namely the “oxindole alkaloids”.
- 12.
In the communication announcing their synthesis of dl-eserethole, the validity of which is beyond any doubt, Julian and Pikl (1935b) state that “ In a series of ten beautiful papers Robinson and his co-workers have described syntheses of compounds which they call “d,l-eserethole” and “d,l-esermethole.” Their “d,l-eserethole” is not the compound (see footnote 13) described in this communication as d,l-eserethole, and the constitution of which can hardly be questioned. We believe that the English authors are in error, that the compound they describe as d,l-eserethole is not the substance, and that we are describing for the first time the real d,l-eserethole”, and summarise that “The “d,l-eserethole” described by other workers in this field is thought to have another constitution than that ascribed to it.” They reinforce this conclusion by the comment that “our product was the real d,l-eserethole and that that of the English chemists must be assigned another constitution” in their communication concerning their complete synthesis of l-physostigmine (Julian and Pikl 1935c). This “very public disagreement between Percy Julian and Robert Robinson, as to the identity of “eserethole” ˮ has been the subject of detailed discussion and analysis in review (Ault 2008; Robinson 2002).
It was, however, recognised by Julian and Pikl (1935b) [and also later in review (Julian et al. 1952b)] that Robert Robinson’s synthetic approach was intact until its final stage [although unlikely (Ault 2008) doubt would appear to have been cast (Kobayashi 1938) upon this] when they noted that (Julian and Pikl 1935b) “The English authors depend upon methylation of their d,l-noreserethole (which seems to be identical with our product.....) with methyl p-toluenesulfonate and at times methyl sulfate. This could well lead to a substance whose structure is represented by [30]. A substance [methyl-eserethole] with this formula assigned to it has been obtained by Hoshino and Kobayashi [1934a – see also 1934b] through methylation by various procedures of dinoreserethole [vide supra]. Its melting point appears to be identical with the “d,l-eserethole” of Robinson and his co-workers and likewise the melting points of the two picrates seem identical”. Indeed, such was reported (King and Robinson 1935) as being corroborated “ by a direct comparison of the specimens”, at which juncture the molecular formula of “methyl-eserethole” was also confirmed as C15H22N2O which contained only two N-methyl groups – ultimately it was shown to have structure 29 (R1=EtO, R2=H, R3=NMe2) (vide supra). Nevertheless, the Oxford group tenaciously refused to accept the unequivocal fact that their synthesis had collapsed at this stage by their statement that “In our opinion the base is structurally identical with eserethole, and it may be a stereoisomeride of this base (see footnote 13). We accept the evidence of Hoshino and Kobayashi [1934a-see also 1934b] (loc. cit.) that the substance is not d,l-eserethole, although the facts leading to this conclusion have not fallen within our experience” (King and Robinson 1935). However, Robert Robinson (see footnote 5) – the future (1947) Nobel Laureate in chemistry (Farber 1963; Saltzman 1993) “for his work on the synthesis of natural products, the alkaloids” (Saltzman 1993) and who has been regarded as “the master of synthesis” (Saltzman 1993) and as “ the king of alkaloid chemistry” (Brossi et al. 1996) – and his group at Oxford were never to reach the synthetic objective of l-PHYSOSTIGMINE. Indeed, this was further manifest by their failure to prepare dl-noresermethole (23, R1=MeO, R2=R3=H, R4=Me) by heating 5-methoxy-1,3-dimethylindole with an excess of ethyleneimine in a sealed tube at 120°C for 3h (King and Robinson 1933) which, perhaps not surprisingly, afforded only unchanged indolic starting material.
Despite the above observations and conclusions, and with reference to the work of Robert Robinson and some of his team [in particular (King et al. 1933b, Robinson and Suginome 1932a, b)], it has been stated (Sumpter and Miller 1954a) that “The first synthesis of the complete ring system of physostigmine was achieved by the preparation of dl-noreserethole [which would appear to have been recognised by Julian and Pikl (1935b) – see also (Henry 1949, Julian et al. 1952b), although Robert Robinson himself occasionally appears not to have realised that he had reached this synthetic destination (Robinson 2002)], which could be methylated to give dl-eserethole” [which is certainly not the case (vide supra) (Robinson 2002)].
- 13.
Robert Robinson and his co-worker FE King sought to assign (King and Robinson 1935) differences (see footnote 14) to variations in molecular geometry at the B/C ring junction of the 1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole system by their statement that “It is relevant to note that Linstead and Meade (1934, 935) have isolated cis-cis- and cis-trans-isomerides of fused dicyclooctanes (two five-membered rings), and we provisionally regard the isomerism of d,l-eserethole-a, mp 38° (synthesised by Julian and Pikl, loc. cit), and our d,l-eserethole-b, mp 80°, as another case of the same kind. The fact that the behaviour of d,l-eserethole-a, and d,l-eserethole-b towards methyl iodide is different is not surprising, because the stereochemical hypothesis closely concerns the configuration of the nitrogen atoms.” (King and Robinson 1935). However, Robinson and King had failed to realise the effect on the eserethole tricyclic system of the aromatic ring A which, by forming a near-planar system with the ring B, thereby restricts the B/C ring junction to a cis fusion (Jackson 1954), (see also Sect. 4). No such restriction is present, of course, in Linstead and Meade’s fused dicyclooctanes and the conclusion to be drawn, therefore, is that “all compounds containing the tricyclic ring system [as in eserethole] can exist in one configuration only, i.e. with the two pyrrolo rings cis to each other, so that for each individual compound only one pair of enantiomorphic forms is possible” (Jackson 1954) and that “All the experimental results so far obtained are in agreement with this hypothesis, and for each compound prepared only one pair of enantiomorphs has ever been obtained, never two pairs as would be predicted on the basis of the two asymmetric centres which the molecule contains.” (Jackson 1954) (see also Sect. 4).
- 14.
Earlier, along with M Liguori (King et al. 1934), they had also suggested that mp discrepancies might have resulted from either dimorphism [“It appears that the substance exists in two crystalline modifications” (King et al. 1934)] or even the “alternative hopeful idea of spontaneous resolution” (King et al. 1934) although this latter “did not survive the experimental test” (King et al. 1934).
- 15.
Nevertheless, King and Robert Robinson (1935) later found it necessary to further state, with understandable reason, that “The identity of the synthetic salts with those derived by degradation of the alkaloid established for the first time the existence of the angle methyl group and gave important support to the view of the constitution of eserine which is now generally accepted. As this fact does not seem to have been recognised by other workers in the field, we venture to draw attention to it”. From the literature cited by the Oxford duo, the “other workers” undoubtedly could have included Julian and Pikl (1935c) who asserted that “The most convincing work on the constitution of eserine, that from Barger’s laboratory [quoting (Stedman and Barger 1925), namely some seven years before the paper by King and Robert Robinson (1932a)] left one important gap unfilled, namely, the synthesis of oxindole derivatives secured as degradation products”. Perhaps, too, the statement by the Oxford pair reflected a frustration at the failure of the synthetic approaches by Robert Robinson and his research group to the actual alkaloid (Sect. 3.1), the successful synthesis of which by Julian and Pikl (1935c) they concomitantly acknowledged (King and Robinson 1935).
- 16.
If, instead of using aluminium chloride alone, a melt of a mixture of aluminium chloride with sodium chloride (5:1 w/w) is employed to effect this type of cyclisation the resulting yields of product may be substantially increased (Sugasawa and Murayama 1958a).
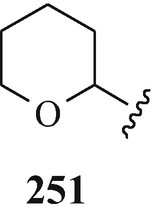
Alternatively, the Stollé approach to the required indolin-2-onic synthetic intermediate could be replaced by the Brunner synthesis (Robinson 1982). Indeed, this is a method of choice for the preparation of 3-alkylindolin-2-ones – affording yields in the region of up to 80% (Robinson 1982) and, furthermore, its use would circumvent the undesired O-dealkylation (Julian and Pikl 1935b; Sugasawa and Murayama 1958a, b) and thereby obviate the necessity for the O-realkylation stage (Julian and Pikl 1935b, Sugasawa and Murayama 1958a, b). An interesting and useful protection of the potential 5-hydroxyl function in a Julian and Pikl type synthesis (Sect. 3.1) involving the use of the tetrahydro-2-pyranyl moiety 251 – apparently affording an acetal, such as the methoxymethyl moiety as used by Kolosov et al. (1953) (see footnote 22) – was developed at the Shanghai Institute of Organic Chemistry (Brossi et al. 1996; Pei et al. 1996; Yu et al. 1994). However, although this latter protecting group affords the best synthesis of the racemic alkaloids, its component chiral centre leads to complications in the synthesis of optically active alkaloids (Brossi et al. 1996; Pei et al. 1996). Perhaps it is of ultimate interest that the Stollé synthesis of 1,3-dimethyl-5-hydroxyindolin-2-one has, in fact, been effected without the need to protect the OH group (Pei et al. 1998; Yu et al. 1994).
- 17.
The reductive cyclisation of, for example 19 [R1=H, R2=(CH2)2NH2] into 23 (R1-R3=H, R4=Me) using sodium in “alcohol” [a Ladenburg reduction (Hino 1961b, Yamada et al. 1963)] requires large amounts of sodium and “alcohol”. This consequently makes the experimental procedure cumbersome and labour-intensive. In addition, the use of large amounts of sodium is dangerous in large-scale reactions and the neutralisation of the base with acid upon work-up results in a large amount of sodium salts (Brossi et al. 1996). These disadvantages, however, were later ameliorated by the introduction of lithium aluminium hydride [which had already been used (Julian and Printy 1949) in stirred ether to effect the reduction of 1-methylindolin-2-ones to 1-methylindoles – other early studies upon the lithium aluminium hydride reduction of oxindoles have been covered in review (Robinson 1917b, 1969)], in either ether or ether/dioxane (Hino 1961b; Hino and Ogawa 1961; Yamada et al. 1963) and in either dioxane or tetrahydrofuran (Brossi et al. 1996; Hendrickson et al. 1962; Hino and Yamada 1963; Sandoz Ltd 1966; Yu and Brossi 1988; Yu et al. 1994) boiling under reflux, as the reducing agent. When proceeding via the formation of the carbamate 19 [R2=(CH2)2 NHCOOMe], these conditions concomitantly result in an ultimate N-methylation to presumably afford the intermediate 19 [R2=(CH2)2NHMe] (Pallavicini et al. 1994), and they can also be employed in the direct reductive cyclisation of the Julian and Pikl nitriles 19 (R2=CH2CN) (Brossi 1990; Sandoz Ltd 1966; Yu and Brossi 1988), thereby obviating the low yields caused by a competing hydrolysis of the nitrile group resulting from the difficulty in excluding moisture from the reaction mixture when using sodium in anhydrous ethanol to effect the reductive cyclisation.
However, the sensitivity of lithium aluminium hydride toward air and the necessary use of highly-flammable peroxide-forming ethers is hazardous, particularly in large scale preparations (Pei and Bi 1994; Pei et al. 1996). Those factors have led to the introduction of sodium dihydride-bis(2-methoxyethoxy) aluminate, known commercially as Vitride, in toluene (Pei and Bi 1994; Pei et al. 1996), as a reducing agent that is chemically equivalent to lithium aluminium hydride but is stable to oxygen, highly soluble in a variety of solvents, easy to work-up, and safe – especially for large scale reductions (Brossi et al. 1996; Pei and Bi 1994; Pei et al. 1996, 1998). Its use can also lead to the isolation of the corresponding 2-indolinols which, via their methiodides can be reacted with a wide variety of nucleophiles to afford products of considerable interest (Pei et al. 1994) (Sect. 3.2.2) (see footnote 3 in Chap. 3).
- 18.
Contrary to the earlier implication (Stedman and Barger 1925) that these reductions, involving zinc and hydrochloric acid (Polonovski 1918, Polonovski and Polonovski 1924e – see also 1924a) and hydrogenation over platinum in glacial acetic acid (Stedman and Barger 1925) must have involved the direct rupture of the C8a-N1 bond, which “seems rather unlikely as this is not activated, e.g. benzylically or allylically”, it has been suggested that they occur via an acid-catalysed ring-C cleavage of, for example, 23, (R2=R3=H, R4=Me) (Jackson and Smith 1964, see also Troxler 1972) to afford the 3H-indolium cation such as 22 and that it is the immonium function thus formed that is reduced (Jackson and Smith 1964). Verification for this arises from the observation that “no reduction occurs in neutral or alkaline solution” (Jackson and Smith 1964). Thus the 1,2,3,3a,8,8a-hexahydro-3a-methylpyrrolo[2,3-b]indole ring system is unaffected under conditions of hydrogenation over a palladium-on-carbon catalyst in methanol (Atack et al. 1989; Brossi 1990; Brzostowska et al. 1992; Yu et al. 1988a, b) and it also explains why the C(8a)-N1 bond in Julian and Pikl’s product is stable to sodium in boiling dry ethanol under reflux.
- 19.
Since phenacetin (acetylphenetidine) (17, R1=EtO, R2=COMe) is one of the so-called “coal tar” analgesics and l-eseroline was some three decades ago (Sect. 10.11) also shown to possess analgesic activity, albeit of an opioid type, curiously it may be considered that this synthesis has, this far, pharmacologically returned whence it commenced.
- 20.
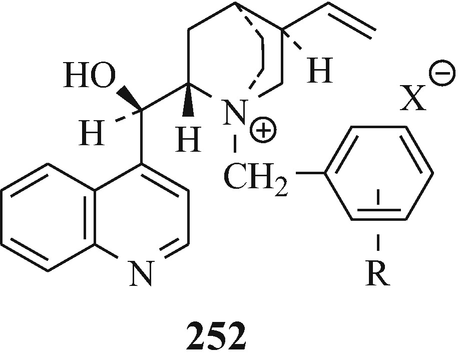
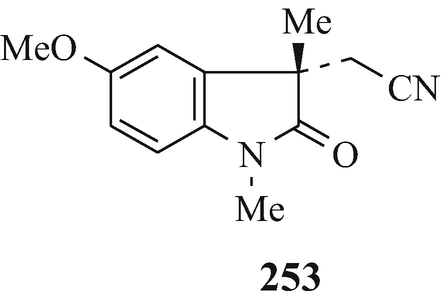
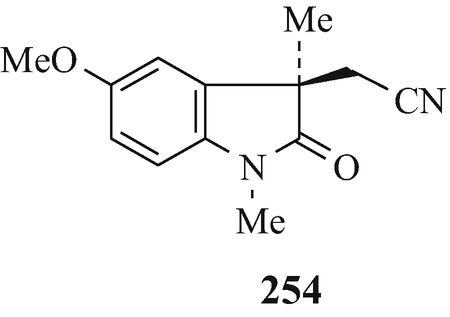
Alkylations such as this can now be effected asymmetrically by phase-transfer catalysis using chiral catalysts. For example, with 19 (R1=MeO, R2=H) in the presence of 252 (R=4-CF3, X=Br; R=3,4-Cl2, X=Br and Cl) under phase-transfer conditions, ees of the S-enantiomer 253 over the R-enantiomer 254 of 72%, 77% and 78%, respectively, were realised at the Chemical Research Department of Hoechst-Roussel Pharmaceuticals Inc. in Somerville, New Jersey, USA (Lee and Wong 1991 ˗ see also Pei et al. 1996). The separation of the mixture of enantiomers thus enriched in one of the two optical isomers can be effected by spontaneous crystallisation (Brossi 1990, 1994; Brossi et al. 1996; Pei and Brossi 1995), a very interesting observation. By thus focussing upon the first prochiral intermediate – namely the nitrile – thereby effected a remarkable improvement in the Julian and Pikl synthesis (Brossi et al. 1996) by affording at an early stage the desired optically pure enantiomer in high yield which obviated the later wasteful formation of undesired enantiomers which would have to be discarded. However, difficulties have arisen using this approach which were overcome by catalytic reduction of the nitrile to the amine which was resolved via formation and a single crystallisation of the diastereoisomeric dibenzoyl-D-tartarates (Lee and Wong 1991).
- 21.
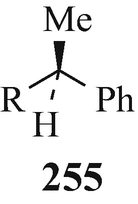
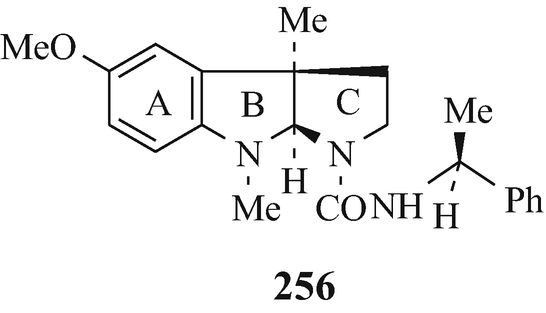
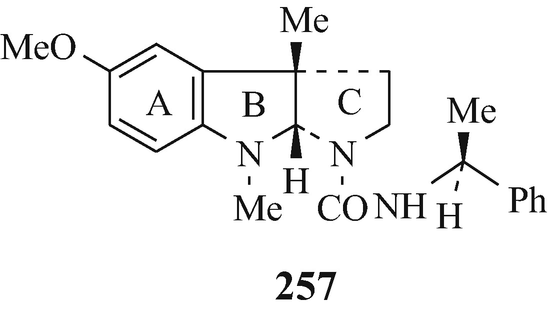
The resolution of racemic amines via their reaction with an optically active acid to afford a mixture of two diastereoisomeric salts that are then separated by fractional crystallisation and from which the optically active amines are subsequently liberated is a widely-employed classical technique. However, sometimes it is either unsuccessful or proves to be difficult and not very practical [despite the use of novel acidic resolving agents which will continue to be of value (Brossi 1994)], with tedious recrystallisations of the salts being required to yield optically pure materials in consequent low yields. Indeed, such is the case with the resolution of dl-N1-noresermethole (23, R1=MeO, R2=R3=H, R4=Me) and dl-eserethole (23, R1=EtO, R2=H, R3=R4=Me) which can thus be separated into their optically pure stereoisomers in only low yields (Brossi et al. 1996; Schönenberger and Brossi 1986; Schönenberger et al. 1986a). However, an alternative method is now available by which the optically active amines can be obtained from their racemates in high yield and of excellent optical pure amines. It is similar in principle to the earlier classical method of resolution, involving the preparation of a mixture of two diastereoisomeric derivatives (but in this case ureas, obtained by reacting enantiomeric secondary amines with optically pure isocyanates), their separation (in this instance chromatographically), and finally the liberation of the optically pure amines. This technique is applicable to the resolution of a wide range of racemic secondary amines including, for example (Brossi, 1985, 1990, 1992, 1994; Brossi et al. 1996; Brossi and Yu 1988a b, Kawabuchi et al. 1988; Schönenberger et al. 1986a, b; Yu et al. 1988a) that of dl-N1-noresermethole (23, R1=MeO, R2=R3=H, R4=Me) in which a stirred solution of the latter in chloroform at 0°C is treated by the dropwise addition of the isocyanate 255 (R=O=C=N) and, after 2h, the solvent is evaporated to leave a mixture of the diastereoisomeric ureas 256 and 257 (a discussion of the stereochemistry of the B/C ring junction in the 1,2,3,3a,8,8a-hexahydro-3a-methylpyrrolo[2,3-b]indole ring system is presented in (see footnote 13)). These are then separated by column chromatography (Brossi 1985; Kawabuchi et al. 1988; Yu and Brossi 1988) [silica gel/CH2Cl2-MeOH (100:1 to 80:1)] and then decomposed by boiling, under reflux and an atmosphere of nitrogen for 2h (Brossi 1985; Schönenberger and Brossi 1986; Schönenberger et al. 1986a, b; Yu and Brossi 1988), with either 1M sodium pentoxide in “pentanol” or sodium butoxide in “butanol”, or in boiling pentan-l-ol or butan-l-ol alone under reflux (Kawabuchi et al. 1988; Schönenberger et al. 1986ab) to afford the component optical isomers of the dl-N1-noresermethole (23, R1=MeO, R2=R3=H, R4=Me) of excellent optical purity and in high yields [besides 255 (R=Me(CH2)4O-CO-NH and Me(CH2)3O-CO-NH, respectively) which can be reconverted into 255 (R = O=C=N) by hydrolysis to afford 255 (R=NH2) and then reaction of this with phosgene] (Schönenberger et al. 1986a, b).
Resolution can also be directly effected chromatographically on a preparative scale on chiral stationary phases. This topic has received comprehensive coverage in reviews (Anton et al. 1994; Brossi 1994; Brossi et al. 1996; Francotte 1994; Francotte and Junker-Buchheit 1992). Specific examples involve the resolution of the Julian and Pikl nitrile 19 (R1=MeO, R2=CH2CN) using a column of microcrystalline cellulose triacetate as the stationary phase and elution with 96% ethanol to afford good yields of the enantiomers 253 and 254 (Brossi et al. 1996; Pei et al. 1996) and a similar analytical chromatographic separation using cellulose-based chiral stationary phases has likewise been affected (Brossi et al. 1996, Lee TBK and Wong 1990) of a variety of indolin-2-ones implicated in the synthesis of l-physostigmine by the Julian and Pikl approach. Furthermore, in another similar application but using 95% ethanol for elution, dl-physovenine (3, R1=MeNHCO, R2=Me, X=O) was separated into its enantiomers 95 (R=MeNHCO, X=O) and 98 (R1=MeNHCO, R2=Me, X=O, n=1) (Yu et al. 1991).
Finally, it is important to note that resolution should be effected as early as possible in the reaction sequence (Pei et al. 1996; Witkop 1998) in order to minimise the effect of the wastage of “an equal amount of the undesired enantioner, which has to be discarded” (Brossi et al. 1996). Indeed, work has “focused on the first prochiral intermediate in the Julian [Julian and Pikl] synthesis, the nitriles [253] and [254], and a great variety of analogues” (Brossi et al. 1996), the synthesis of which can be asymmetrically directed using phase-transfer catalysts (see footnote 20) and which can be separated by chromatography on a microcrystalline cellulose triacetate column (Brossi et al. 1996; Pei et al. 1996) (vide supra).
- 22.
“The yield from this reaction was greatly increased when the reaction mixture was stirred continuously throughout the boiling period” (Dale and Robinson 1970), a similar O-demethylation having also been affected when the reaction mixture was boiled under reflux with stirring under an atmosphere of nitrogen (Hill and Newkome 1969). However, attempted O-demethylation of homoesermethole [racemic (98, R1=R2=Me, X=NMe, n=2)] to produce the corresponding phenol by heating with “anhydrous aluminium chloride in gasoline (b.p. 70-77o)” was unsuccessful, as was the same attempted cleavage of the ether bond by heating with aniline hydrochloride and by treatment of either the base or its hydrobromide with either hydrobromic acid or hydrochloric acid and with variation in the concentration of acid, reaction temperature and length of heating – apparently, “demethylation proceeded only under condition which led to the formation of an inseparable mixture of substances” (Kolosov et al. 1953). Perhaps O-demethylation in this case might be possible using boron tribromide in dichloromethane which was later (Luo et al. 1990; Robinson et al. 1987, 1988; Schönenberger et al. 1986a; Shishido et al. 1986b; Yu and Brossi 1988) used to effect such transformations in good yields. However, in the absence of this possibility, Kolosov et al. (1953) introduced the methoxymethyl moiety as a protecting group since the resulting methoxymethylether “was, like all acetals, stable enough in alkaline, and readily decomposed in acidic media” from under which conditions the desired product, homoeseroline [racemic (98, R1=H, R2=Me, X=NMe, n=2)], could be isolated. Similar properties are inherent in the acetal function of the product when using the tetrahydro-2-pyranyl moiety 251 as the protecting group (see footnote 16) and which can be easily removed with 2N-hydrochloric acid (Brossi et al. 1996; Pei et al. 1996; Yu et al. 1994).
- 23.
Percy Lavon Julian (1899–1975)
It is poignant to note that the funding of this research [(Julian and Pikl 1935abc), Sect. 3.1] appears to reflect then current social attitudes prevalent in the USA in that the concluding paragraph of the final paper announcing the development of the synthesis – claimed to be “undoubtedly the most significant chemical research publication to come from De Pauw” (Anon 1999) – reads “In acknowledging a generous grant from the Rosenwald Fund, the senior author respectfully dedicates this finished project to the memory of Julius Rosenwald, who has made possible innumerable cultural contributions on the part of young negroes to his country’s civilization” (Julian and Pikl 1935c). Indeed, when he had the pleasure of meeting with this most delightful man at his Julian Research Institute (vide infra) in Chicago on 22nd June 1972, it became apparent to the author that Dr Julian, as a result of his being an Afro-American [he had been born on 11th April 1899 in Montgomery, Alabama, the second generation out of slavery (Anon 1999; Borman 1993)] had, during the early period of his life, for many years experienced and been the victim of the racial segregation and prejudice that was then practised within the USA, a fact that was later verified during a televised biography of him (The Percy Julian Biography Project 2002). His father, James Sumner Julian, was a railway clerk and that his mother, Elizabeth Lena Julian (née Adams), was a school teacher resulted in the young Julian – in a society where only 41% of black children attended school beyond the age of ten years and more than 50% of the black population was illiterate – staying on in school and doing well (Borman 1993).
Another acknowledgement of gratitude, but in this instance appended to each of the four publications (Julian et al. 1934; Julian and Pikl 1935abc) leading to the l-physostigmine synthesis, is directed toward Dr WM Blanchard, Dean of the College of Liberal Arts and Senior Professor of Chemistry at DePauw University. The significance of this appreciation not only lies in the fact that it was Blanchard who first imbuded Julian with his predilection for chemistry whilst he was an undergraduate at DePauw – where, in the Autumn of 1916, aged 17, he had been accepted as a sub-freshman (at which juncture his family, in order to enable them to give him their full support, moved with him to Greencastle, Indiana), and from where, in 1920, he graduated BA as valedictorian (ie at the top of his class), despite, in addition to his regular university courses, his having to take remedial classes at a nearby high school – but later created the opportunity for him, after he had left Howard University under a cloud of university politics that were probably racially motivated (vide infra), to do the research at DePauw that led to his celebrated first synthesis of l-physostigmine (Sect. 3.1) (see footnotes 24 and 25).
Julian’s career received help from two North American philanthropic organisations, namely the General Education Board (that was financed by the Rockefeller family) and the Julius Rosenwald Fund (Rosenwald was the chairman of the Sears Roebuck catalog sales company), both organisations which played significant roles between 1900 and 1940 in improving the educational opportunities for Afro-Americans by supporting and stimulating the construction of large numbers of appropriate schools across the South of the USA. Although these benefits were too late to help Percy Julian, both these organisations also made selective awards in support of up-and-coming black scientists and amongst whom he was to be included. However, in the meantime after his graduation from DePauw (vide supra), and because of the lack of future employment opportunities, he spent two years as instructor in chemistry at Fisk University, in Nashville, Tennessee and in 1922, with the help from Blanchard at DePauw (vide supra), under the auspices of an Austin Fellowship he commenced graduate studies at Harvard University from where, after one year, he graduated MA and finished, yet again, at the top of his class but on the campus of which race became a major issue and once again raised its ugly head, with his application for a teaching assistantship being repeatedly rejected lest Southern students were offended by having a “Negro” teacher (Borman 1993). Consequently, Julian, supporting himself with minor Fellowship positions and odd jobs – such as stoking furnaces – outside the university, remained as a researcher at Harvard until 1926 when he moved to West Virginia State College for Negros as professor of chemistry and then, two years later, to the predominately black Howard University in Washington DC as associate professor of chemistry and where he quickly rose to full professor and the head of the chemistry department. It was from here in 1929 that he at last threw off the yolk of racism and, frustrated in his efforts to pursue doctoral studies in the USA, moved as a Rockefeller Foundation Scholar, with the associated financial support from the General Education Board (vide supra), to the University of Vienna where, under the direction of Ernst Späth, he resumed graduate studies and began his doctoral research in the area of the chemistry of natural products, specifically on the indole alkaloids of Corydalis cava (Späth and Julian 1931), a plant that grows in the Vienna Wood.
Subsequent upon ultimate graduation as PhD (Vienna) in 1931 as another part of his doctoral research [see, for example (Julian 1931)] he returned to Howard University as a Post-doctoral Research Fellow financed by the Julius Rosenwald Fund and accompanied by Josef Pikl, a friend from his days in Vienna (Witkop 1998). After two years at Howard, internal politics forced them to leave, at which juncture, once again through the efforts of William Blanchard (vide supra), Julian, accompanied by his Julius Rosenwald Post-doctoral Research Fellowship, returned to DePauw and it was here in the Minshall Laboratory in 1935, in collaboration with Josef Pikl, who was then working as an assistant in chemistry (Anon 1999), that he completed the research that resulted in the first total synthesis of l-physostigmine (Sect. 3.1) and (see footnotes 24 and 25) and that established his reputation, at the age of thirty-six years, as a world-renowned organic chemist and an internationally recognised published researcher. However, despite these accomplishments and the recommendations and support of Dean WM Blanchard (vide supra) and DePauw President G Bromley Oxnam, the university’s Board of Trustees rejected Julian’s application for a faculty teaching position – sadly DePauw was not ready for a black professor – and the University of Minnesota likewise rejected him. Thus frustrated in his attempts to secure an academic position, Julian turned his attention to industry and was offered a research position at the Institute of Paper Chemistry at Appleton, Wisconsin. However, the Institute’s officials became aware of an Appleton statute that forbade “housing of a Negro overnight” and, consequently, “Julian’s career at the Institute ended before it began” (Borman 1993).
However, a door was soon to be opened which was to lead to a long-term and very successful industrial career and came about as a result of an attempt by Julian – whilst still working at DePauw University (Anon 1999, Witkop 1998) – to isolate geneserine (Chap. 6) from Calabar beans which ultimately led to his isolation, from the component oil of this botanical source, of the hydrate of stigmasterol (175, R=H) (Chap. 9). Since the most common source of this sterol was soy bean oil, Julian sought five gallons of this oil from The Glidden Company in Chicago. It was at this juncture that a white vice-president at Glidden’s, WJ O’Brien, being aware of Julian’s excellent scientific reputation and his rebuff at Appleton (vide supra), offered him the position as Assistant Director of Research at Glidden’s Soya Product Division, an offer that Julian gladly accepted in 1936. Thus began a remarkable industrial career in the vibrant city of Chicago where he was to spend the rest of his life and which has been superbly reviewed by one of Julian’s friends – over a time span of almost sixty years which went back to the first synthesis of l-physostigmine (Sect. 3.1), Bernhard Witkop (1980, 1998). The numerous patents that emanated over eighteen years from his soy bean protein research work at The Glidden Company, Soya Product Division, and which led after the first four years to the Division becoming the company’s most profitable single entity, produced many successful products for Glidden, including a paper coating and a fire-retardant foam which saved many lives from its wide use in Aerofoam by US troops during World War II to fight and extinguish petroleum and oil fires. His first patent (Julian et al. 1940), which resulted from a fortuitous isolation of stigmasterol from soy bean oil, ultimately led to synthetic work [see, for example, (Julian et al. 1949, 1950, 1951, 1956a)] which produced in large quantities and at low cost progesterone [see (Witkop 1998)] and cortisone, following the epochal joint Nobel Prize-winning (in 1950) discovery by Edward C Kendall, Philip S Hench and Tadeus Reichstein (Sourkes 1966) that the latter product reversed the symptoms of rheumatoid arthritis. Eventually, Julian became research director in Glidden’s soya products division, and also concurrently acted as manager of their fine chemicals division and research director of its Durkee Famous Foods division (Borman 1993).
In 1953 Glidden’s decided to cease all work on steroids and sold Julian’s patents to Syntex and Pfizer, two of the companies involved in the “Great Steroid Wars of the 1950s”. Consequently, in that same year Julian resigned from The Glidden Company to establish his own laboratory, Julian Laboratories, Inc, in Franklin Park, Illinois which specialised in steroid chemistry and the production of therapeutical steroids from Mexican yams. In 1961 he sold this remarkably successful company to Smith, Kline and French Laboratories for more than 2.3 million US dollars whilst remaining as president on a generous salary – later he repurchased one of the laboratories from Smith, Kline and French and established a non-profit making research organisation in Chicago called the Julian Research Institute. From here, via an arrangement with the graduate center at the University of Illinois, he directed the doctoral research of post-graduate students (Borman 1993).
Throughout Percy Julian’s remarkable multifaceted professional career runs an unbroken series of publications relating to various aspects of the chemistry of indoles. This begins – as part of his doctoral research at the University of Vienna (vide supra) – with an application of that doyen of the indole syntheses, namely that discovered by Emil Hermann (Farber 1970; Lucier 1993) Fischer (Robinson 1982), to methyl 3,4-dimethoxyphenylethyl ketone phenylhydrazone to furnish 3-(3,4-dimethoxy)benzyl-2-methylindole (Julian 1931). This was followed in turn by a sole contribution – and, in addition, a “non-indolic paper” (Julian and Passler 1932) – from Howard University (vide supra) (Julian and Pikl 1933) and from DePauw University by the celebrated quartet (Julian et al. 1934, Julian and Pikl 1935a, b, c) which ultimately afforded l-physostigmine (Sect. 3.1) and by (Julian et al. 1935 and Julian and Pikl 1935d). Then a series of papers, several concerning yohimbine (Julian et al. 1948a, b, c; Julian and Magnani 1949), were published jointly from DePauw University (vide supra) and from the Research Laboratories of The Glidden Company, Soya Products Division (vide supra) (Julian et al. 1948a) and from the latter organisation alone (Julian et al. 1945, 1948b, c, 1953, Julian and Magnani 1949; Julian and Printy 1949, 1953) and from Julian Laboratories, Inc (vide supra) (Julian et al. 1956b, c). As well as his work on the synthesis of yohimbine alkaloids, he also effected investigations into the metabolism of the amino acid tryptophan (Anon 1999, Julian et al. 1935, 1956b, c, Witkop 1978, 1983). Also from the Research Laboratories of The Glidden Company, Soya Products Division (vide supra) he co-authored (Julian et al. 1952b) a review on “THE CHEMISTRY OF INDOLES” [which incorporates references to some of his as then unpublished studies (Julian 1933, 1952; Julian and Magnani 1952; Julian and Passler 1952; Julian and Printy 1952; Julian et al. 1952a, c)] and which, alongside that by Sumpter and Miller (1954b), provided not only invaluable contemporary comprehensive converages of the subject but still remain as classical texts of considerable utility and interest. Furthermore, an illustration captioned “Julian discusses ongoing research project at Julian Research Institute [vide supra] in 1968” (Borman 1993) shows to his left a blackboard liberally festooned with indole-related chemical structures – clearly, “once an indole-man, always an indole-man!”
Further biographical data on Percy Lavon Julian – grandson of slaves, chemistry teacher, teacher and researcher, researcher and administrator, pioneer in the chemical synthesis of therapeuticals, successful industrial research director and manager, millionaire entrepreneur and researcher, the first Afro-American to achieve a supervisory research position within a major American corporation (vide supra) and to serve as a faculty member of a non-Negro college in the USA (vide infra), and civil rights steadfast advocate – who overcame prejudice, blatant discrimination in employment, and racist threats including two attacks by bombers and arsonists (fortunately without injury to either himself or any member of his family) on his home in Chicago where he and his family had been the first Afro-Americans to purchase a home in all-white Oak Park, Illinois – can be found in (Anon 1999; Borman 1993; DeKruif 1946; The Percy Julian Biography Project 2002; Witkop 1980, 1998).
Toward the end of what must be described as an eventful and extraordinary life, in 1974 Julian became ill and it was necessary for him to scale back his activities. He died, aged 76 years, on 19th April 1975.
From Julian’s research in “pure” and “applied” chemistry emanated, respectively, 51 publications from 1931 to 1969 and 117 patents variously issued in Australia, France, Germany, Great Britain and the USA (Witkop 1998). He received 19 honorary degrees – whilst alive or posthumously, in 1973 he was elected to the National Academy of Sciences, and was paid a rare philatelic tribute when he was honoured, in the US Postal Services black heritage commemorative series, by the release on 30th January 1993 of a new 29 cent stamp for nationwide sale (Borman 1993). He also played an active role in the National Association for the Advancement of Colored People, The Chicago Urban League and the Mental Health Association of Greater Chicago and was a trustee at six colleges and universities, included in which was the Board of Trustees of DePauw University to which he was appointed in 1967 –by a “school that had declined to hire him as a faculty member more than 30 years before” (vide supra) (Borman 1993). At this later juncture, “planning began for a new science building, which would replace the 65-year-old Minshall Laboratory [where the celebrated first synthesis of l-physostigmine had been effected (Sect. 3.1)], and construction commenced in 1968. The Science and Mathematics Center was dedicated in September 1972, with Percy Julian giving the dedication address, ‘Science and the Good Life of Man’ ” and “Following Julian’s death, DePauw University named the Percy L. Julian Science and Mathematics Center in his honor” (Anon 1999). Also at DePauw, an annual lecture series in his honour was, in 1993, in its seventeenth year, and a scholarship established by his family has provided help and support to many of that university’s students (Borman 1993).
The author’s memory returns to 22nd June 1972 (vide supra) when, at the Julian Research Institute in Chicago (vide supra), he had the great pleasure and privilege to meet and spend a most delightful day with Dr Julian, a person who he fondly remembers with much respect and great affection.
- 24.
In fact, the soundness and versatility of this synthetic approach were soon – indeed, at its outset – recognised when, included in their earliest publication (Julian et al. 1934) – announcing the conversion of 19 (R1=R2=H) into 23 (R1=R2=H, R3=R4=Me), Julian and Pikl (along with Doyle Boggess) stated that “Since, moreover this nitrile [19 (R1=H, R2=CH2CN)] is obtained in 90% yield and [19 (R1=R2=H)] is a very cheap substance, the way is opened to a ready synthesis of the drug itself and even of homologs, containing other groups in the 3-position of the indole nucleus” (Julian et al. 1934) and confidently predicted that “We hope soon to communicate the complete synthesis of the drug itself by this same procedure” (Julian et al. 1934). This goal was ultimately referred to (Hino 1961b) as a “brilliant synthesis of physostigmine” and, furthermore, as recently as 1998 it has been stated (Witkop 1998) that “Although there have been more than half a dozen subsequent syntheses of physostigmine [vide infra], Julian’s classical approach, with some recent modifications and improvements (see footnote 16, 17, 20, 21, 22), is still the best route to the synthetic alkaloid....”, an opinion with which the author concurs (see footnote 25).
The recent improvements (Yu et al. 1994) (see footnote 16, 17, 20, 21, 22) to the Julian and Pikl synthesis were mostly developed at the National Institutes of Health in Bethesda, Maryland, USA, at the Shanghai Institute of Organic Chemistry, China, at Georgetown University in Washington DC, USA and at the Chemical Research Department of Hoechst-Roussel Pharmaceuticals, Inc in Somerville, New Jersey, USA, and all of which have been the subject of review (Brossi et al. 1996, Greig et al. 1995a). Indeed, using these improvements, the Julian and Pikl approach to the total synthesis of the ring system of l-(3a,S–cis)- physostigmine and its optical isomer has been achieved on a large and technical scale (Pei et al. 1996, Brossi 1992), as has that of phenserine (98, R1=PhNHCO, R2=Me, X=NMe, n=1) (Sect. 10.7.2) (Brossi et al. 1996). Furthermore, applications of the synthesis to that of 3a–aryl substituted 1,2,3,3a,8,8a-hexahydropyrrolo[2,3–b]indoles proceed via the 3-cyanomethylation of the appropriate 3-arylindolin-2-ones as has been exemplified using 3-phenylindolin-2-one [interestingly in the presence of sodium hydride instead of sodium ethoxide as used by Julian and Pikl (1935a, b, c) – the alklation of indolin-2-one at the 3-position has also been well-investigated by others (Hino 1961a)] (Sandoz Ltd 1966) and 5-methoxy-l-methyl-3-phenylindolin-2-one which was prepared via a Stollé synthesis starting from reaction of 4-methylaminophenol (17, R1=OH, R2=Me) [an interesting and useful circumvention of the need to protect the hydroxyl function, as also utilised in another instance (Yu et al. 1994)] with 2-bromo-2-phenylacetyl chloride) (Pei et al. 1998).
- 25.
The synthesis’s most recent accolade was commemorated by its designation, conferred by the American Chemical Society, as a National Historic Chemical Landmark (Anon 1999). A plaque marking this event was presented to DePauw University on 23rd April 1999, during the university’s celebration of the centennial anniversary of Percy Julian’s birth, and reads (Anon 1999):-
NATIONAL HISTORIC
CHEMICAL LANDMARK
SYNTHESIS OF
PHYSOSTIGMINE
DePauw University
Greencastle, Indiana
1935
In 1935, in Minshall Laboratory, DePauw alumnus Percy L. Julian (1899–1975) first synthesised the drug physostigmine, previously only available from its natural source, the Calabar bean. His pioneering research led to the process that made physostigmine readily available for the treatment of glaucoma. It was the first of Julian’s lifetime of achievements in the chemical synthesis of commercially important natural products.
American Chemical Society
April 23, 1999
- 26.
Similar in mechanistic principle to this ring closure and the related earlier studies is the reaction between Koshland’s protein reagent, 2-hydroxy-5-nitrobenzyl bromide (a reagent which reacts specifically with tryptophan residues in a protein chain) and tryptophan which was shown (McFarland et al. 1969), almost exclusively on the basis of 1H-nmr spectroscopic data, to afford a mixture of two isomeric products, 258 and 259 – although no structure was proposed for a minor reaction product. It is obvious that the formation of ring C in 258 and 259 results from the electrophilic attack of the 2-hydroxy–5–nitrobenzyl cation at the 3-position of the indole nucleus, followed by ring closure involving nucleophilic addition of the nitrogen atom of the 3-substitent at the C-2 atom of the 3H-indolium cation so formed (Robinson 1917b 1971). However, it would appear that the amino group involved in this cyclisation does not need to be free since N-acetyltryptamine and N-acetyl-L-tryptophan methyl ester also afford similar structurally-related products (Spande et al. 1968). That from the former reaction was acetylated to yield a triacetyl derivative for which structure 260 (R=H), rather than the ring-C opened 3H-indole 261 (R=H) was preferred (Spande et al. 1968) although structural confirmation by X-ray crystallography is awaited (Robinson 1971; Spande et al. 1968). Three products were isolated from the latter reaction followed by acetylation. The major product, after separation by chromatography, appears to have structure 260 (R=COOMe) from the comparison of its uv – and 1H-nmr – spectral properties with these of 260 (R=H) (Spande et al. 1968) – the structure of the minor reaction products remain to be determined (Robinson 1971; Spande et al. 1968).
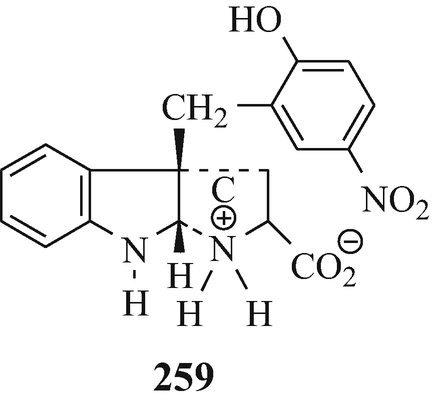
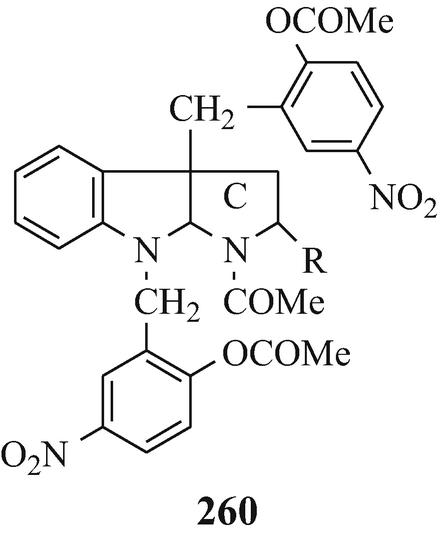
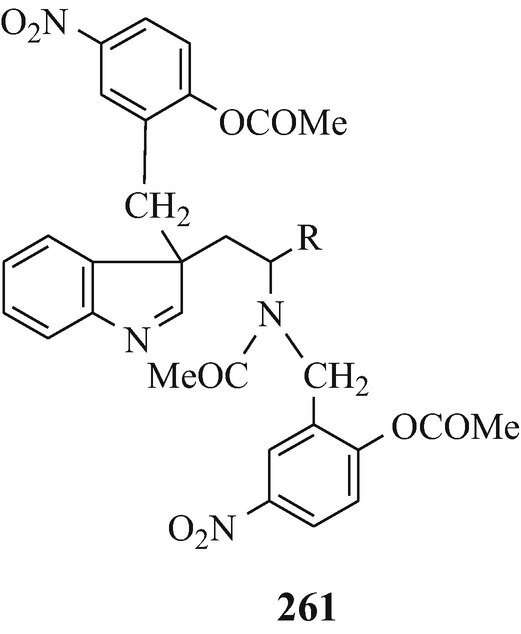
A mechanism similar to that involved in the formation of 258 and 259 (vide supra) must be operational in the formation of compound 262 from the reaction between N-acetyltryptamine and 3,3-dimethylallyl bromide (Casnati et al. 1969), but now with the initial stage involving an electrophilic attack by a 3,3-dimethylallyl moiety (Casnati et al. 1969; Robinson 1971). These above reactions, together with other mechanically-related transformations, have already been subjected to review (Fontana and Toniolo 1976; Hino and Nakagawa 1988).
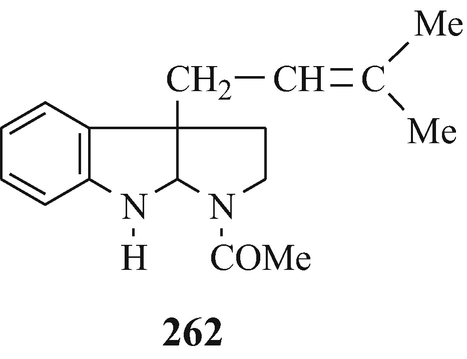
The above cyclisations leading to 258 and 259 are analogous to those that have been long established as following the 3-alkylation of suitably activated tryptamines (Sect. 3.1). Furthermore, similar ring closure also follows electrophilic addition at the indolic 3-position of tryptamines and tryptophans by the diazonium salt derived from 2-methoxy-4-nitroaniline (“Fast Red B”) (Fontana and Toniolo 1976), a proton (see footnote 4 in Chap. 3), positive halogen [such as Cl⨁ derived from t-butyl hypochlorite (see footnote 4 in Chap. 3), Br⨁ derived from N-bromosuccinimide (see footnote 4 in Chap. 3) and I⨁ derived from iodine azide (Ikeda et al. 1979)], and of singlet and positive oxygen (vide infra). These latter pair provide a synthetic route to the 1,2,3,3a,8,8a-hexahydro -3a-hydroxypyrrolo[2,3-b] indole system – that occurs naturally in brevianamide E (228, R=CMe2CH=CH2) (Birch and Russell 1972; Birch and Wright 1970; Hino and Nakagawa 1988; Nakagawa et al. 1975; Sammes, 1975), flustraminols A and B (Christophersen 1985b); hunteracine [Herbert 1983(b); Husson 1983; Nakagawa et al. 1975], okaramine (232) (Hayashi et al. 1989 and sporidesmin A 227) (Fridrichsons and Mathieson 1965; Nakagawa et al. 1975) – which has been effected by photooxidation of tryptamines, and to the corresponding 2-carboxy- and 2-carbomethoxy analogues by either peroxyacetic acid oxidation [in water using one mole of preformed oxidant at 0–5 oC for 24h (Savige 1975)] or photoxidation of either tryptophans or their methyl esters (Fontana and Toniolo 1976; George and Bhat 1979; Saito et al. 1977), respectively. In two further interesting examples of these transformations, compound 263, formed (Hino and Nakagawa 1988) via a Meisenheimer rearrangement and which is structurally analogous to l-geneserine (Sect. 6.1), was isolated as one of the products resulting from the Rose Bengal sensitised photoxidation of N1-methyltryptamine (George and Bhat 1979; Nakagawa et al. 1975; Saito et al. 1977) and the photooxidation of tryptophol afforded a near quantitative yield of 264 (R=OOH) which is structurally analogous to l-physovenine (Sect. 3.1) and which readily decomposed under a variety of conditions to afford a mixture of products including 264 (R=OH) (George and Bhat 1979; Saito et al. 1977).
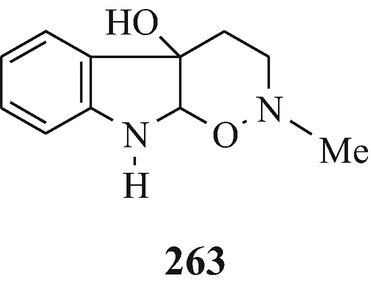
- 27.
In view of the results obtained by Hoshino et al. (1934), the statement (Taylor 1966) – unfortunately made without reference to a primary source – that “Experimentally, however, treatment of 5-ethoxytryptamine with ethyl magnesium iodide followed by reflux with methyl iodide gave instead the methyl derivative of the ring open tautomer” [namely 29 (R1=EtO, R2=H, R3=NMe2)] would appear to be erroneous. Indeed, it has probably emanated from confusion with the formation of this product, namely “methyl-eserethole”, by methylation of compounds already containing the 5-ethoxy-1,2,3,3a,8,8a-hexahydro-3a-methylpyrrolo[2,3-b]indole system, such as 23 (R1=EtO, R2-R4=H, R2=R4=H, R3=Me and R2=R3=H, R4=Me) (see footnote 12).
- 28.
This claim was later shown to be erroneous since the product gave a picrate, small yellow prisms from “alcohol”, mp 180-181°C (King et al. 1934) which is clearly different from the picrate – orange crystals from “alcohol”, mp 150°C (Kolosov et al. 1953) [a later observation reported orange-yellow dice from ethanol, mp 162-163°C (Sugasawa and Murayama 1958a, b)] – of dl-esermethole (23, R1=MeO, R2=H, R3=R4=Me) that was independently and unequivocally synthesised using Julian and Pikl’s approach involving as the final step the reductive cyclisation of 19 [R1=MeO, R2=(CH2)2NHMe] using sodium in boiling either “absolute butyl alcohol” (Kolosov et al. 1953) or “dehyd. ethanol” (Sugasawa and Murayama 1958a, b).
- 29.
This product gave a picrolonate as light brown, tiny, crystalline aggregates from “alcohol”, mp 220°C (decomp) (King and Robinson 1932b) while the picrolonate of dl-noresermethole (33, R1=MeO, R2=R3=H, R4=Me) that was to be unequivocally synthesised by an application of Hoshino and Kobayashi’s route (vide supra) has mp 221°C (Hoshino et al. 1934). Similarly it afforded a deep red picrate as long rhombic plates from methanol, mp 159°C (King and Robinson 1932b) which would appear to be identical with the picrate [reddish-orange rhomboidal prisms from “alcohol”, mp 162-163° (King et al. 1934) and dark red prisms from ethanol, mp 163-164°C (Robinson 1965a, b)] of dl-noresermethole that was later, respectively, obtained by the Oxford group (King et al. 1934) (vide supra) and subsequently as an intermediate in a synthesis of dl-eseramine (Sect. 4.2.1) via a Julian and Pikl type approach involving the reductive cyclisation of 19 (R1=MeO, R2=CH2CN) using sodium in dry ethanol boiling under reflux (Robinson 1917b, 1965a, b).
- 30.
In view of the results from investigations referred to elsewhere (Sect. 6) and (see footnote 18), it might be expected that the intermediate 44 (R=H) would undergo opening of its tetrahydropyrrolo ring C to afford 43 (R=Me, X=p-Tosyl). The latter is of a special interest in that it represents a monoquaternary derivative of a diamine in which it is the least basic nitrogen atom that is quaternised and therefore it might be expected to undergo methyl group transfer from its least basic to a more basic nitrogen atom. Such a transfer could be intramolecular to afford 29 (R1=EtO, R2=H, R3=⊕NHMe2 p-Tosyl⊖), basification of which would give “methyl-eserethole” (29, R1=EtO, R2=H, R3=NMe2). Alternatively, since the reaction was effected (King et al. 1933a) by mixing solutions of equimolar quantities of dl-noreserethole (23, R1=EtO, R2=R3=H, R4=Me) (in dry benzene) and methyl p- toluenesulphonate (in ethyl acetate), the methyl migration might be intermolecular, namely from 43 (R=Me, X=p-Tosyl) to 23 (R1=EtO, R2=R3=H, R4=Me) – since both these species would have co-existed in the reaction medium – to afford 29 (R1=EtO, R2=H, R3=NHMe) and 44 (R=H) –and this cycle could then continue – and the former product could undergo quaternisation with methyl p-toluenesulphonate to afford 29 (R1=EtO, R2=H, R3=⊕NHMe2 p-Tosyl⊖) which upon basification would yield “methyl-eserethole”.
In addition, under these reaction conditions it is likely that dl-eserethole (23, R1=EtO, R2=H, R3=R4=Me) [produced via the intermolecular quaternisation of 23 (R1=EtO, R2=R3=H, R4=Me (vide supra)] and methyl p-toluenesulphonate would have co-existed in the reaction medium and could have thereby reacted to yield 44 (R=Me), opening of the tetrahydropyrrolo ring-C with subsequent intermolecular transfer of the methyl group from the quaternary nitrogen atom of the 3H-indolium cation thus formed to the more basic nitrogen atom of another molecule of dl-eserethole (23, R1=EtO, R2=H, R3=R4=Me) thus giving “methyl-eserethole” (29, R1=EtO, R2=H, R3=NMe2) and 44 (R=Me) – and this other cycle could, once again, then continue.
It is now suggested that since both these above sequences involve the presence of methyl p-toluenesulphonate, they could explain the lack of formation of “methyl-eserethole” and the almost quantitative recovery of unchanged dl-eserethole as the only product resulting from compound 44 (R=H) in the investigations by Jackson (1954) which are, unfortunately, not accompanied by relevant experimental details that are far from unequivocal from the quoted literature. Perhaps a clarification of this problem may be sought by an investigation of the product(s) resulting from a repeat of the study by Jackson (1954) (vide supra) in which dl-eserethole methyl p-toluenesulphonate (44, R=H) is “ treated under exactly the same conditions” as were employed for the methylation of dl-noreserethole (King et al. 1933a) (see also King et al. 1934; King and Robinson 1935) but now in the presence of either ethyl p-toluenesulphonate or 14C-methyl p-toluenesulphonate .
- 31.
The trivial name echiboline has been derived (Fritz and Fischer 1964) from the observation that this tetracyclic ring system is present in the alkaloid echitamine (216) (Robinson 1963a; see also Hamilton et al. 1962) (see footnote 9) and its positional numbering has been defined to be as shown in 89 (R1-R3=H2, X=H2, n=2) (Fritz and Fischer 1964). A facile synthesis of this parent compound has been effected (Fritz and Fischer 1964; Robinson 1971) by cyclisation of 4a-(2-aminoethyl)-1,2,3,4,4a,9a-hexahydro-4aH-carbazole, formed by treatment with hydrazoic acid of 4a-(2-carboxyethyl)-1,2,3,4,4a,9a-hexahydro-4aH-carbazole which was one of the two products obtained by Fischer indolisation of the phenylhydrazone of 71[R3+R4=(CH2)4, R5=COOH, n=1]. This reaction sequence as might that starting with the Fischer indolisation of 87 (n=2) (Sect. 3.2), also be applicable to the synthesis of 6-hydroxyechibolines with antinociceptive activity (Sect. 10.11). Another synthetic route to such latter products could be attempted – using an approach based upon that developed by Hoshino and Kobayashi, with their occasional co-workers (Sect. 3.1) – by reacting 1,2,3,4-tetrahydro-6-methoxycarbazole with either ethyl or methyl magnesium iodide followed by N-methylaziridine –a nucleophile suggested (Onaka 1971) for then future use in this type of reaction – with subsequent hydrolytic “work-up”. This should afford 89 (R1=MeO, R2=H, R3=Me, X=H2, n=2) and ultimately lead to 6-hydroxyechibolines in which the N(9)-substituent could be other than a methyl group.
- 32.
It is interesting that whereas reduction of 6-methoxy-9-methyl-11-oxoechiboline (see footnote 31) (89, R1=MeO, R2=Me, R3=H, X=O, n=2) using lithium aluminium hydride in boiling tetrahydrofuran under reflux afforded 6-methoxy-9-methylechiboline (89, R1=MeO, R2=Me, R3=H, X=H2 n=2), an attempt to effect a similar reduction of a suspension of 89 (R1=MeO, R2=Me, R3=H, X=O, n=2) in boiling diethyl ether under reflux, in which it is only very slightly soluble, was unsuccessful and when, under these conditions, a Soxhlet apparatus was employed to effect dissolution, an unexpected reductive ring scission occurred along with reduction of the carbonyl group to afford 4a-(2-aminoethyl)-6-methoxy-9-methyl-1,2,3,4,4a,9a-hexahydrocarbazole (Cox and Robinson 1988). Since it is well-established (see footnote 18) that reductive cleavage of the Ph-N-C-N system only occurs under acidic conditions, probably via acid-catalysed ring opening to the corresponding 3H-indolium cations (Jackson and Smith 1984), the possible mechanism for the ring opening in this instance was discussed although the reason(s) for the relationship between the nature of the reaction product and the reaction conditions remained unclear (Cox and Robinson 1988).
- 33.
Indeed, it has been claimed (Kutney 1977) that the Fischer indole synthesis (Robinson 1982) is “the most versatile and widely applied reaction particularly in the syntheses of natural alkaloids”. For examples, it has been employed in the initial stage of the synthesis of strychnine (Kutney 1977; Woodward et al. 1954, 1963), as early stages in those of harmalan and tryptophan (Julian et al. 1952b), in various intermediary stages of those of dl-dasycarpidone, dl-3-epi-dasycarpidone, dl-uleine and dl-3-epi-uleine (Jackson et al. 1969; Kutney 1977), ellipticine (Hewlins et al. 1984; Sainsbury 1977; Stillwell 1964), harmaline (Sumpter and Miller 1954b) and the β-carboline alkaloids (Kutney 1977), in the antepenultimate stage of syntheses of dl-aspidospermine (Ban et al. 1965; Stork and Dolfini 1963 – see also Ban et al. 1969; Inoue and Ban 1970; Kutney 1977; Robinson 1982; Stevens 1977), in the penultimate stage of the synthesis of dl-quebrachamine (Stork and Dolfini 1963 – see also Ban et al. 1969; Kutney 1977; Stevens 1977) and as the final stages in two (Ikezaki et al. 1969; Sallay 1967 – see also Augustine and Pierson 19690; Kutney 1977) syntheses of dl-ibogamine, in that of cinchonamine (Ch’ang-pai et al. 1958, 1960 – see also Robinson 1982) [which, furthermore, had earlier (Witkop 1950) been converted into quinamine using dilute peracetic acid] and the ring-system of yohimbine (Clemo and Swan 1946; Julian and Magnani 1949; Kutney 1977). Furthermore, its applications in Borsche-type syntheses (Robinson 1982), which have also been claimed [Joule 1983(h)] as affording the best method of preparing the ring A-substituted dimethylcarbazoles for use in the Saxton route to the synthesis of ellipticine and its analogues, has led to the carbazole (so-called) (see footnote 4) alkaloids (Kapil 19710; Kutney 1977), in (so-called) abnormal “advanced” Fischer indolisations (Ishii 1980, 1981) to the naturally-occurring 6-isoprenylindoles (Ishii and Murakami 1975 – see also Ishii 1981), and have also been involved in the syntheses of 5-hydroxytryptamine, 5-hydroxytryptophan and 3-indolylacetic acid, together with many of their analogues and homologues (Julian et al. 1952a, b, c; Robinson 1982). It has also be utilised in some of the synthetic approaches that have been made to the 3a-alkyl-3,3a,8,8a-tetrahydro-2H-furo[2,3-b]indole ring system that is present in physovenine (Sect. 3.2.3) (Robinson 1982).
- 34.
The difference in behaviour of this system in l-physovenine by comparison with that reported (Bardsley 1961) for compound 264 (R=Me) (Hoshino and Shimodaira 1935) which shows indoline absorption in neutral ethanol, benzenoid absorption in dilute acid and a mixture of benzenoid and 3H-indolium cation absorption in concentrated acid is probably because of the absence in this latter compound of the MeNHCOO group that is present at the 5-position of l-physovenine [cf (Joule and Smith 1962)] (Robinson 1964a). Moreover, and interestingly, it has been concluded (Jackson and Smith 1964) that “It is clear that formation of these indolium salts has been, and will continue to be, a useful diagnostic test for the presence of the Ph-N-C-N and Ph-N-C-O systems in newly discovered indole alkaloids. The two systems can be distinguished from each other by the fact that only the former can undergo quaternisation in dilute acids, and hence give the characteristic small hypsochromic shift first observed by [GF] Smith”. Indeed, such examinations played a major role not only in the elucidation of the structure of l-physovenine as 3 (R1=MeNHCO, R2=Me, X=O) (Robinson 1964a) (Sect. 3.1) but also in establishing the structure of the product resulting from the treatment of 69 (R1=Et, R2=⨁NMe3 IƟ, X=O) with boiling caustic soda as 98 (R1=Et, R2=Me, X=O, n=1), the uv spectrum of which in ethanol indicated the indoline nucleus, in dilute ethanolic hydrochloric acid was characteristic of a mixture of indoline and 3H-indolium cation chromophores, and in concentrated hydrochloric acid had pure 3H-indolium cation absorption (Longmore 1969; Longmore and Robinson 1966) (Sect. 3.2.2).






















































References
Abramovitch RA (1958) Tryptamines, carbolines, and related compounds, part IV. Attempted synthesis of the physostigmine ring system. Can J Chem 36:354–357
Abramovitch RA, Muchowski JM (1960) Tryptamines, carbolines, and related compounds, Part VI. Stereochemistry of the Michael addition of ethyl malonate to 1-cyclohexene cyanide. Can J Chem 38:557–566
Adolf W, Hecker E (1975) On the irritant and cocarcinogenic principles of Hippomane mancinella. Tetrahedron Lett:1587–1590
Ahmed M, Robinson B (1967) The ultraviolet spectra of the 5-methoxy-3,3-dimethyl and 1,3,3-trimethyl-3H-inolium cations. J Chem Soc (B):411–413
Anon (1945) Quantitative organic microanalysis based on the methods of Fritz Pregl, 4th English edn, completely revised by Grant J (ed) J & A Churchill, London, pp 146–160
Anon (1999) A national historic chemical landmark. Synthesis of physostigmine. DePauw University, Greencastle, Indiana. April 23, 1989 (American Chemical Society, Division of the History of Chemistry and The Office of Public Outreach, 1155 Sixteenth Street, N W Washington, DC 20036)
Anton K, Eppinger J, Frederiksen L, Francotte E, Berger TA, Wilson WH (1994) Chiral separations by packed-column super- and subcritical fluid chromatography. J Chromatogr 666:395–401
Armit JW, Robinson R (1925) CCXI. Polynuclear heterocylic aromatic types. Part II. Some anhydronium bases. J Chem Soc 127:1604–1618
Ashimori A, Matsuura T, Overman LE, Poon DJ (1993) Catalytic asymmetric synthesis of either enantiomer of physostigmine. Formation of quaternary carbon centers with high enantioselection by intramolecular heck reactions of (Z)-2-butenanilides. J Org Chem 58:6949–6951
Atack JR, Yu Q, Soncrant TT, Brossi A, Rapoport SI (1989) Comparative inhibitory effects of various physostigmine analogues against acetyl– and butyrylcholinesterases. J Pharmacol Exp Ther 249(1):194–202
Attitulah R, Bardsley WG, Smith GF, Lehey N (1966) 4th Internat. Symp. on Chem of Nat Prod (IUPAC), Stockholm, June abstr 2B-7, 84 [this is summarised in (Gorman et al. 1971)]
Augustine RL, Pierson WG (1969) The synthesis of dl-deethylibogamine. J Org Chem 34:1070–1075
Ault A (2008) Percy Julian, Robert Robinson and the identity of eserethole. J Chem Educ 85(11):1524–1530
Auterhoff H, Hamacher H (1967) Color reactions of eserine. Arch Pharm (Weinheim) 300:849–856 [Chem Abs (1968) 68: 6207y]
Bacchi A, Pelizzi G, Redenti E, Delcanale M, Amari G, Ventura P (1994) Geneserine hydrochloride. Acta Cryst C50:1126–1130
Ban Y, Sato Y, Inoue I, Nagai (née Seo) M, Oishi T, Terashima M, Yonemitsu O, Kanaoka Y (1965) Total synthesis of aspidospermine. Tetrahedron Lett 6(27):2261–2268
Ban Y, Akagi M, Oishi T (1969) Synthesis and stereochemistry of octahydro-4H-pyrrolo[3,2,1-i,j]quinolin-9(2H)-one. Tetrahedron Lett (25):2057–2062
Bardsley WG (1961) Part 1. Dehydrobufotenine. Part 2. The indoline NCO chromophore. MSc Thesis, Victoria Manchester (see in particular 24–26)
Barger G (1936) From physostigmine to Prostigmin. Festschrift Emil C Barell, Basel, pp 7–17
Barrow CJ, Cai P, Snyder JK, Sodlock DM, Sun HH, Cooper R (1993) Win 64821, a new competitive antagonist to substance P isolated from an Aspergillus species: structure determination and solution conformation. J Org Chem 58:6016–6021
Benešová V, Samek Z, Herout V, Šorm F (1969) Plant substances XXIX isolation and structure of two new indole alkaloids from Riccardia sinuata (hook ) Trev. Collect Czechoslov Chem Commun 34:1807–1809
Bentley KW (1957) The physostigmine group in the alkaloids, vol 1. Interscience, New York/London, pp 158–161
Bersch HW (1950) C-Methylation of dihydroberberine with formaldehyde. Arch Pharm 283:193–303 [Chem Abs (1951) 45:2009i]
Bickel H, Schmid H, Karrer P (1955) Zur kenntnis des fluorocurins und maracurins. Helv Chim Acta 38:649–669
Bild N, Hesse M (1967) 196. Notiz über die massenspektren von N-oxiden. Helv Chim Acta 50:1885–1892
Birch AJ, Russell RA (1972) Studies in relation to biosynthesis-XLIV. Structural elucidations of brevianamides-B, −C, −D and –F. Tetrahedron 28:2999–3008
Birch AJ, Wright JJ (1970) Studies in relation to biosynthesis XLII. The structural elucidaton and some aspects of the biosynthesis of the brevianamides -A and – E. Tetrahedron 26:2329–2344
Boardman B, Robinson B (1973) Unpublished observations [quoted in (Longmore and Robinson B 1973)]
Boggess D (1934) MA Thesis, DePauw [quoted as ref 1 in (Julian et al. 1934)]
Boit H-G (1961) Ergebnisse der alkaloid-chemie bis 1960. Akademie-Verlag, Berlin [quoted as ref 1004a in (Hesse M 1964) – see also (Holmes 1879)]
Borman S (1993) Black chemist Percy Julian commemorated on postage stamp. Chem Eng News February 1:9–12
Boyd-Barrett HS (1932) Experiments on the synthesis of physostigmine (eserine). Part IV. The Plancher rearrangement of indole derivatives. J Chem Soc:321–325
Boyd-Barrett HS, Robinson R (1932) Experiment on the synthesis of physostigmine (eserine), Part III. Synthesis of desethoxydehydroeseretholemethine. J Chem Soc:317–321
British Pharmacopoeia (1968) The pharmaceutical press, London
Brossi A (1985) Further explorations of unnatural alkaloids. J Nat Prod 48:878–893
Brossi A (1990) Physostigmine. In: Bioactive alkaloids. 4. Results of recent investigations with colchicine and physostigmine. J Med Chem 33: 2311–2319
Brossi A (1992) Fifteen years of research of bioactive alkaloids. Med Res Rev 12(1):1–26
Brossi A (1994) Chiral drugs:synopsis. Med Res Rev 14:655–691
Brossi A (1997) Alkaloids in medicine in encyclopedia of human biology, vol 1. 2nd edn. Academic,pp 243–247
Brossi A, Pei X (1998) Biological activity of unnatural alkaloid enantiomers. In: Cordell GA (ed) The alkaloids chemistry and biology. Academic, San Diego/London/Boston/New York/Sydney/Tokyo/Toronto. 50: chapter 3, 109–139 and refs therein cited
Brossi A, Yu Q (1988a) Preparation of carbamates related to (+)-physostigmine as cholinergic agents.US Pat Appl US 166825 15 Jul 1988, Appl 04 Mar 1988; 21pp Avail NTIS Order no PAT-APPL-7-166 825 [Chem Abstr (1988) 109: 190632x]
Brossi A, Yu Q (1988b) Preparations of carbamates related to (−)-physostigmine as cholinergic agents. US Pat Appl US166824 15 Jul 1988, Appl 04 Mar 1988; 22pp Avail NTIS Order no PAT-APPL-7-166 824 [Chem Abs (1988) 109:190633y]
Brossi A, Pei X, Greig NH (1996) Invited review phenserine, a novel anticholinesterase related to physostigmine: total synthesis and biological properties. Aust J Chem 49:171–181
Bruncko M, Crich D, Samy R (1994) Chemistry of cyclic tautomers of tryptophan : formation of a quaternary center at C3a and total synthesis of the marine alkaloid (+)-ent-debromoflustramine B. J Org Chem 59:5543–5549
Brunner K (1898a) Ueber die E Fischer’sche, aus methylketol und jodmethyl darstellbare base. Ber 31:612–615
Brunner K (1898b) Ueber die E Fischer’sche, aus methylketol and jodmethyl darstellbare base. Ber 31:1943–1949
Brustier V (1926) Contribution à l’étude spectrographique de l’absorption des rayors ultra-violets par les alcaloides et les glucosides. Bull Soc Chim France 39:1527–1543 and in Thèse doctorat Médecine, Toulouse, 1926, and in CR Acad Sci, 1930, 190:499 [quoted in Brustier V (1932) Les specres d’absorption ultra-violets des alcaloides. Application de leur étude a la connaissance de la structure moléculaire de ces substances, a l’analyse chimnique, a la pharmacie, a la toxicologie. Chim Ind 27(5):1007–1016]
Brzostowska M, He X, Greig NH, Rapoport SI, Brossi A (1992) Phenylcarbamates of (−)-eseroline (−)-N1-noreseroline and (−)- physovenol: selective inhibitors of acetyl and of butyrylcholinesterase. Med Chem Res 2:238–246
Budzikiewicz H, Djerassi C, Williams DH (1964) Structure elucidation of natural products by mass spectrometry. Volume I: alkaloids. Holden-day Inc., San Francisco, London, Amsterdam chapter 10. Physostigmine and related alkaloids. pp 162–172
Carlé JS, Christophersen C (1979) Bromo-substituted physostigmine alkaloids from a marine bryozoa Flustra foliacea. J Am Chem Soc 101:4012–4013
Carlé JS, Christophersen C (1980) Marine alkaloids. 2. Bromo alkaloids from the marine bryozoan Flustra foliacea. Isolation and structure elucidation. J Org Chem 45:1586–1589
Casnati G, Francioni M, Guareschi A, Pochini A (1969) Insertion of isoprene units into indole systems. Tetrahedron Lett 10:2485–2487
Ch’ang-pai C, Evstigneeva RP, Preobrazhenskii NA (1958) The synthesis of the alkaloid cinchonamine. Proc Acad Sci USSR 123:927–928
Ch’ang-pai C, Evstigneeva RP, Preobrazhenskii NA (1960) Synthesis of the natural alkaloid (+)- cinchonamine. J Gen Chem USSR 30:2066–2068
Chemnitius F (1927) Zur darstellung des physostigmins. J Prakt Chem 116:59–64
Christison R (1855) On the properties of the ordeal-bean of Old Calabar, West Africa. Monthly J Med, 20 (3rd series, 11): 193–204 [for a somewhat abridged presentation of this article, see (Christison 1854–1855)]
Christophersen C (1983) Marine indoles. In: Marine natural products. Academic, Chapter 5, pp 259–285
Christophersen C (1985a) Secondary metabolites from marine bryozoans. A review. Acta Chem Scand B39:517–529
Christophersen C (1985b) Marine alkaloids. In: Brossi A (ed) The alkaloids chemistry and pharmacology, vol 24. Academic, Orlando/San Diego/New York/London/Toronto/Montreal/Sydney/Tokyo, Chapter 2, Bryozoan alkaloids, section III. E, for subsequent communications see, for example, refs A and 431
Clayton E, Reed RI (1963) Electron impact and molecular dissociation – XV. The mass spectra of physostigmine and some related compounds. Tetrahedron 19:1345–1357
Clayton E, Reed RI, Wilson JM (1962) Electron impact and molecular dissociation – VII. The calycanthus alkaloids. Tetrahedron 18:1495–1501
Clemo GR, Swan GA (1946) The constitution of yohimbine. Part I. J Chem Soc:617–621
Cordell GA (1981) Physostigmine and related compounds. In: Introduction to alkaloids a biogenetic approach. Wiley, New York/Chichester/Brisbane/Toronto, pp 584–591
Cordell GA (1983) The bisindole alkaloids, chapter 11. In: Saxton (ed) (1983), pp 543(a), 583(b), 715(c), 716(d)
Cox B, Robinson B (1988) The reduction of 6-methoxy-9-methyl-11-oxoechiboline using lithium aluminium hydride. J Heter Chem 25:271–272
Coxworth E (1965) Alkaloids of the Calabar bean. In: Manske RHF (ed) The alkaloids chemistry and physiology, vol 8. Academic, New York/London, Chapter 2, pp 27–45
Coyne WE, Paterson GR (1961) Degradation studies on physostigmine and structural studies on rubreserine. Can Pharm J 94:45–49
Cozzi PG, Palazzi C, Potenza D, Scolastic C, Sun WY (1990) Total synthesis of (±) – pseudophrynamine A. Tetrahedron Lett 31(39):5661–5664
Creasey WA (1983) Pharmacology, biochemistry, and clinical applications of the monoterpenoid alkaloids, chapter 14. In: Saxton (ed) (1983), pp 783–829(a), 788(b), 789(c), 799(d), 806(e), 808–816(f), 816–819(g)
Crich D, Pavlovic AB, Samy R (1995) The chemistry of cyclic tautomers of tryptophan: total synthesis of (+)-(ent)-pseudophrynaminol. Tetrahedron 51(23):6879–6384
Crooks PA, Robinson B, Meth-Cohn O (1976) The 13C-nuclear magnetic resonance spectra of physostigmine and related compounds. Phytochemistry 15:1092–1093
Dale FJ (1969) The synthesis and anti-acetylcholinestease activity of (+)-physostigmine and (+)-physovenine. MSc Thesis, Victoria Manchester
Dale FJ, Robinson B (1970) The synthesis and anti-acetylcholinesterase activities of (+)-physostigmine and (+)-physoverine. J Pharm Pharmacol 22:889–896
Daly JW, Garraffo HM, Pannell LK, Spande TF, Sererini C, Erspamer V (1990) Alkaloids from Australian frogs (myobatrachidae): pseudophrynamines and pumilotoxins. J Nat Prod 53(2):407–421
Dalziel JM (1948) The useful plants of West Tropical Africa. The Crown Agents for the Colonies, 4.Millbank,Westminster, London, S.W.1 – under the authority of the Secretary of State for the Colonies, 256 [an appendix to the 1st edition of (Hutchinson and Dalziel 1958)]
de Jesus AE, Steyn PS, van Heerden FR, Vleggaar R, Wessels PL, Hull WE (1983) Tremorgenic mycotoxins from Penicillium crustosum: isolation of penitrems A-F and the structure elucidation and absolute configuration of penitrem A1. J Chem Soc (Perkin I), 1847–1856
DeKruif P (1946) The man who wouldn’t give up. Reader’s Digest Magazine, August, pp 113–118
Dolby LJ, Furukawa S (1963) A novel ring expansion of 3-indoleacetamides. J Org Chem 28:2512–2514
Döpke W (1963) Alkaloide aus Calabar-bohnen. Naturwissenschaften 50:713
Dowley P, Gillard RD, Robinson B (1966) Unpublished observations [quoted in (Longmore and Robinson B 1969a, Robinson B 1968)]
Ehrenberg A (1893) Über alkaloide der calabarbohnen. Verh Ges Deut Naturforch Ärzte 102–104 [Chem Centr (1894), 6, Band 2:439]
ElAzab AS, Taniguchi T, Ogasawara K (2000) An expedient route to the Calabar bean alkaloids (−)-physovenine and (−) – physostigmine. Org Lett 2(18):2757–2759
Ellestad GA, Mirando P, Kunstmann MP (1973) Structure of the metabolite LL-5490β from an unidentified Aspergillus species. J Org Chem 38(24):4204–4205
Ellis S (1943) Studies on physostigmine and related substances IV. Chemical studies on physostigmine breakdown products and related epinephrine derivatives. J Pharmacol 79:364–372
Fales HM, Lloyd HA, Milne GWA (1970) Chemical ionization mass spectrometry of complex molecules II alkaloids. J Am Chem Soc 92:1590–1597
Farber E (1963) Nobel prize winners in chemistry (1901–1961) (new and revised edition) [1947 Robert Robinson (1886– )]. Abelard – Schuman, London/New York/Torronto, pp 198–202
Farber E (1970) Fischer, Emil Hermann. In: Gillispie CC (ed) Dictionary of scientific biography, vol 5. Charles Scribner’s Sons, New York, pp 1–5
Ficken GE, Kendall JD (1961) Diazaindenes and their quaternary salts. Part II. The cyclisation of isopropyl methyl ketone 3-pyridylhydrazone. J Chem Soc:584–588
Fletcher G (1968) MSc Thesis, Bath [quoted in (Fletcher and Davies 1968)]
Fletcher G, Davies DJG (1968) The effect of pH and sodium metabisulphite on the stability of physostigmine sulphate solutions to heat and ionising radiation. J Pharm Pharmacol 20(S1):108S–113S
Flippen-Anderson JL, Deschamps JR, Brossi A, Greig NH (2002) 2-Hydroxybenzoic acid salt of physostigmine. Acta Cryst E58:0853–0855
Fontana A, Toniolo C (1976) The chemistry of tryptophan in peptides and proteins. Fortschr Chem Org Naturst 33:309–449. and refs therein cited
Francotte E (1994) Contribution of preparative chromatographic resolution to the investigation of chiral phenomena. J Chromatogr 666:565–601
Francotte E, Junker-Buchheit A (1992) Preparative chromatographic separation of enantiomers. J Chromatogr 576:1–45
Fraser TR (1863) On the characters, actions, and therapeutical uses of the ordeal bean of Calabar (Physostigma venenosum, Balfour). Edinburgh Med J 9. 36–56, 123–132, 235–248
Freise FW (1936a) A hitherto unknown physostigmine-containing plant. Pharm Zentralhalle 77:378–379. [Chem Abs (1937) 31: 15524]
Freise FW (1936b) Pharm Zentralhalle 77:378 [quoted as ref 1030 in (Hesse M 1964)]
Fridrichsons J, Mathieson AML (1965) The structure of the methyene dibromide adduct of sporidesmin at −150°C. Acta Cryst 18:1043–1052
Fridrichsons J, Mackay MF, McL MA (1967) The molecular structure of hodgkinsine, C33H38N6. Tetrahedron Lett [36] 8:3521–3524
Fridrichsons J, Mackay MF, McL MA (1974) The absolute molecular structure of hodgkinsine. Tetrahedron 30:85–92
Fritz H, Fischer O (1964) Tetracyclische α-amino-indole. Eine neue verbindungsklasse mit ring-ketten-tautomerie sowie die synthese eines modifizierten c-curains-III. Tetrahedron 20:1737–1753
Fritz H, Stock E (1970) Synthese eines tetracyclischen α-aminoindolins mit den strukturmerkmalen deseserins über eine Fischersche indolsynthese mit fixierung des β-stickstoffs. Tetrahedron 26:5821–5829
Gaddum JH (1954) Anticholinesterases. The history of work on anticholinesterases. Chem Ind:266–268
Gannon WF, Benigni JD, Suzuki J, Daly JW (1967) The synthesis of dehydrobufotenine. Tetrahedron Lett 8:1531–1533
George MV, Bhat V (1979) Photooxygenations of nitrogen heterocycles. Chem Rev 79:447–478. and refs therin cited
Gorman AA, Hesse M, Schmid H, Waser PG, Hopff WH (1971) Bisindole alkaloids in a specialist periodical report. The alkaloids. The chemical Socity, London 1: chapter 14, pp 200–209
Grandberg II (1974) Indolylalkylamines from arylhydrazines and γ- or δ-halocarbonyl compounds (review). Chem Het Comp (Engl Transl) 10:501–510 – see also (Grandberg and Tokmakov 1975)
Grandberg II (1983) Tryptamines and related structures from γ-and δ-halogeno-carbonyl compounds and arylhydrazines. J Org Chem USSR 19:2135–2147. and refs therein cited
Grandberg II, Ivanova TA, Yaryshev NG (1970) Indoles XII. An investigation of ring-chain tautomerism in compounds of the eserine series. Chem Het Comps 6(9):1191–1198
Grant IJ, Hamor TA, Robertson JM, Sim GA (1962) The structure of chimonanthine. Proc Chem Soc:148–149
Grant IJ, Hamor TA, Robertson JM, Sim GA (1965) The structure of chimonanthine: X-ray analysis of chimonanthine dihydrobromide. J Chem Soc:5678–5696
Greig NH, Pei X, Soncrant TT, Ingram DK, Brossi A (1995a) Phenserine and ring C hetero-analogues: drug candidates for the treatment of Alzheimer’s disease. Med Res Rev 15(1):3–31
Greig NH, Sambammurti K, Yu Q, Brossi A, Bruinsma GB, Lahiri DK (2005a) An overview of phenserine tartrate,a novel acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Curr Alzheimer Res 2:281–290
Gunn C, Carter SJ (1965) Cooper and Gunn’s dispensing for pharmaceutical students, vol 12, 11th edn. Pitman Medical Publishing Co Ltd, London, p 13
Hall ES, McCapra F, Scott AI (1967) Biogenetic-type synthesis of the calycanthaceous alkaloids. Tetrahedron 23:4131–4141
Hamilton JA, Hamer TA, Robertson JM, Sim GA (1962) The structure of echitamine: X-ray analysis of the methanol solvate of echitamine bromide. J Chem Soc:5061–5075
Harley-Mason J (1948) The structure of adrenochrome and its reduction products. Experientia 4:307–308
Harley-Mason J, Jackson AH (1954a) Hydroxytryptamines, Part I. Bufotenine, 6-hydroxybufotenine and serotonin. J Chem Soc:1165–1171
Harley-Mason J, Jackson AH (1954b) Hydroxytryptamines, Part II, a new synthesis of physostigmine. J Chem Soc 54:3651–3654
Hart NK, Johns SR, Lamberton JA, Summons RE (1974) Psychotridine, a C55H62N10 alkaloid from Psychotria beccarioides (Rubiaceae). Aust J Chem 27:639–646
Hayashi H, Takiuchi K, Murao S, Arai M (1989) Structure and insecticidal activity of new indole alkaloids, okaramines A and B, from Penicillium simplicissimum AK-40. Agric Biol Chem 53(2):461–469
Hellberg H (1949) Stability of eserine solutions – especially eye drops. Svensk Farm Tid 53:658–666. [Chem Abs (1950) 44: 3671h]
Hemsworth BA, West GB (1970) Anticholinesterase activity of some degradation products of physostigmine. J Pharm Sci 59:118–120
Hendrickson JB, Rees R, Göschke R (1962) Total synthesis of the calycanthaceous alkaloids. Chimonanthine. Proc Chem Soc:383–384
Henry TA (1949) Alkaloids of Calabar bean. In: The plant alkaloids, 4th edn. J&A Churchill Ltd, London, pp 539–553. [and also earlier editions such as (Henry 1924)]
Herbert RB (1983) Stuctural and biosynthetic relationships, Chapter 1. In: Saxton (ed) (1983), 24(a), 27(b), 37(c)
Herzig J, Lieb H (1918a) Zur kenntnis des eserins. Monatsh Chem 39:285–292
Herzig J, Lieb H (1918b) Zur kenntnis des eserine. Monatsh Chem 39:285–292 [Chem Abs (1919), 13:421; J Chem Soc (Abs) (1918) 114:504]
Herzig J, Meyer H (1897) Weitere bestimnungen des alkyls und stickstoff. Monatsh Chem 18:379–392
Hesse O (1867) Ueber physostigmine. Liebig’s Ann Chem Pharm 141:82–86
Hesse M (1981) Alkaloid chemistry, vol 26. Wiley, New York/Chichester/Brisbane/Toronto, pp 170–172
Hesse M, v Phillipsborn W, Schumann D, Spiteller G, Spiteller-Friedmann M, Taylor WI, Schmid H, Karrer P (1964) Die strukturen von C-fluorocurin C. mavacurin und pleiocarpamin. Helv Chim Acta 47:878–911
Hewlins MJE, Oliveira-Campos A-M, Shannon PVR (1984) Synthetic approaches to ellipticines and other derivatives and analogues of 6H-pyrido[4,3-b]carbazole. Synthesis April 1984:289–302
Higashide E, Asai M, Ootsu K, Tanida S, Kozai Y, Hasegawa T, Kishi T, Sugiro Y, Yoneda M (1977) Ansamitocin, a group of novel maytansinoid antibiotics with antitumour properties from Nocardia. Nature 270:721–722
Hill RK, Newkome GR (1969) The absolute configuration of physostigmine. Tetrahedron 25:1249–1260
Hinman RL, Lang J (1960) The position of protonation and basicities of indoles. Tetrahedron Lett (21):12–15
Hino T (1961a) Synthetic approaches to calycanthaceous alkaloids. II. A synthesis of 1,1′-dimethyl-3,3′-bis(2-aminoethyl)-3,3′-bioxindole. Chem pharm bull (Tokyo) 9:979–988 (ref 5–7 cited therein)
Hino T (1961b) Synthetic approaches to the calycanthaceous alkaloids. III. Reductive cyclization of 3,3′-bioxindoles. (1). A formation of 5,7-dimethyl-11b,11c-bis(2-dimethylaminoethyl)-5,5a,6a,7,11b,11c-hexahydrofuro[2,3-b:5,4-b']diindole. Chem Pharm Bull (Tokyo) 9:988–995
Hino T, Nakagawa M (1988) Chemistry and reactions of cyclic tautomers of tryptamines and tryptophans. In: Brossi A (ed) The alkaloids, vol 34. Academic, New York, Chapter 1, pp 1–75
Hino T, Ogawa K (1961) [quoted as a footnote on p 991 in (Hino 1961b) and as ref 32b in (Coxworth 1965) ˗ details have been reported in (Yamada S et al. 1963)]
Hino T, Yamada S (1963) Total synthesis of (±)-folicanthine. Tetrahedron Lett (25):1757–1760
Hino T, Tanaka T, Matsuki K, Nakagawa M (1983) Synthesis of (±-)-flustramine B, a marine alkaloid. Chem Pharm Bull (Toyko) 31:1806–1808
Hodson HF, Smith GF (1956) The structure of folicanthine. Chem Ind:740–741
Hodson HF, Smith GF (1957) The structure of folicanthine. Part II. J Chem Soc:1877–1880
Hodson HF, Robinson B, Smith GF (1961) Chimonanthine, a new calycanthaceous alkaloid. Proc Chem Soc 465
Holmstedt B (1972) The ordeal bean of Old Calabar: The pageant of Physostigma venenosum in medicine. In: Swain T (ed) Plants in the development of modern medicine. Harvard University Press, Cambridge, 303–360 (this article is superbly illustrated)
Honda K, Goto M, Kurahashi M, Akizawa T, Yoshioka M, Butler jun VP (1991) Structure of bufothionin. Acta Cryst C47:1506–1508
Hoshino T (1932a) New synthesis of indolenines and that of the eserine nucleus. Abstr Japan Chem Lit 6:390–391 [Chem Abs (1933) 27:291]
Hoshino T (1932b) New method of preparing indolenines and the synthesis of the so-called eserine nucleus. Proc Imp Acad (Tokyo) 8:171–174. [Chem Abs (1932) 26: 4814)]
Hoshino T, Kobayashi T (1934a) Synthetical experiments on eserine. III. Synthesis of d-and l-eserethol methiodides. Proc Imp Acad (Tokyo) 10:564–567 [Chem Abs (1935) 29:18294]
Hoshino T, Kobayashi T (1934b) Synthetic experiments on eserine. II. Proc Imp Acad (Tokyo) 10:99–102 [Chem Abstr (1934) 28:34116]
Hoshino T, Kobayashi T (1935) Synthese des dl-eseräthole. Synthetische versuche über eserin. IV. Synthesen in der indol-gruppe. XIII. Liebig’s Ann Chem 520:11–19
Hoshino T, Kobayashi T (1936) Synthesis of l-eserethole. Proc Imp Acad (Tokyo) 11:416–417 [Chem Abs (1936) 30:59964)]
Hoshino T, Shimodaira K (1935) Synthese des bufotenins und über 3-methyl-3-β-oxyäthyl-indolenin. Liebig’s Ann Chem 520:19–30
Hoshino T, Shimodaira K (1936) Über die synthese des bufotenin-methyl-äthers (5-methoxy-N-dimethyl-tryptamin) und bufotenins (synthesen in der indol-gruppe XV). Bull Chem Soc Japan 11:223–224
Hoshino T, Tamura K (1932) Synthetische versuche in der indol-groppe. X. Über die synthese des eserin-ring systems. Liebig’s Ann Chem 500:42–52
Hoshino T, Kobayashi T, (in part) Kotake Y (1934) Synthetische versuche über eserine. III. Synthese von d- and l-eseräthol-jodmethylat. Synthesen in der indol-gruppe. XII. Liebig’s Ann Chem 516:81–94
Hoshino T, Kobayashi T, Shimodaira K (1935) Synthetical experiments on eserine. IV. Syntheses of dl-eserethole and bufotenine. Proc Imp Acad (Tokyo) 11:192–194 [Chem Abs (1935) 29: 58387]
Husson A-P (1983) The Strychnos group, Chapter 7. In: Saxton (ed) (1983), p 310
Ikeda M, Ohno K, Katsura M, Chun M-W, Tamura Y (1979) Reactions of tryptophols and Nɑ -acetyltryptamines with iodine azide. Formation of 3a-azido-3,3a 8, 8a-tetrahydro-2H-furo- and 3a-azido-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indoles. J Chem Soc (Perkin 1) 3061–3063
Ikezaki M, Wakamatsu T, Ban Y (1969) The total synthesis of (±)-ibogamine. J Chem Soc Chem Commun 88–89 [quoted in (Robinson B 1982)]
Inoue I, Ban Y (1970) Studies on the synthesis of Aspidosperma and related alkaloids. Part II. A synthetic approach to the C–21 oxygenated Aspidosperma alkaloids. J Chem Soc (C) 602–610
Irvine FR (1961) Woody plants of Ghana with special reference to their uses. Oxford University Press, London, pp 402–404, 792
Ishii H (1980) Abnormal Fischer indolization and its development to a new synthetic method. Yuki Gosei Kagaku Kyokaishi 38 (7):693–706 [Chem Abs (1981) 94:15480w]
Ishii H (1981) Nucleophilic displacement of the methoxy group in abnormal Fischer indolization of 2-methoxyphenylhydrazones. Acc Chem Res 14:275–283
Ishii H, Murakami Y (1975) Fischer iondolization [sic] and its related compounds – X application of the advanced Fischer indolization of a 2-methoxyphenylhydrazone derivative to syntheses of some naturally occurring 6-subsitiuted indoles. Tetrahedron 31:933–938
Iwasa T, Harada S, Sato Y (1979) Physostigmine. Jpn Kokai Tokkyo Koho 79, 62,390 (Cl. Cl2013/00), 19 May 1979, Appl.77/127,900, 24 Oct 1977 [Chem Abs (1979) 91:106603t]
Iwasa T, Harada S, Sato U (1981) Miticidal antibiotics C-8030 B, C, D, and physostigmine produced by Streptomyces pseudogriseolus subsp. Iriomotensis subsp. NOV. Takeda Kenkyushoho, 40(1/2):12–26 [Chem Abs (1981) 95:148651v] [see also J Takeda Res Lab 40:12 – as used and quoted as ref 9 in (Murao and Hayashi 1986)]
Jackson AH (1954) Synthetic studies in the tryptophan and tryptamine fields. PhD dissertation, Cambridge
Jackson AH, Smith AE (1964) The protonation of tryptamine derivatives in acidic media. J Chem Soc 5516–5517 and refs therein cited [see also (Robinson B 1969)]
Jackson A, Wilson ND, Gaskell AJ, Joule JA (1969) The synthesis of (±)-dasycarpidone, (±)-3-epi-dasycarpidone, (±)-uleine and (±)-3-epi-uleine. J Chem Soc (C):2738–2747
Jaeger RH, Lewis PME, Robinson R (1974) Reduction of brazilein. Occurrence of reductive coupling. Tetrahedron 30:1295–1300
Jensen H, Chen KK (1936) The chemical identity of certain basic constituents present in the secretions of various species of toads. J Biol Chem 116:87–91
Jobst J, Hesse O (1864) Ueber die bohne von Calabar. Liebig’s Ann Chem Pharm 129:115–121
Joule JA (1983) The sarpagine-ajmaline-akuammiline group, chapter 5. In: Saxton (ed) (1983), pp 219(a), 220(b), 245(c), 248(d), 249(e), 250(f), 251(g)
Joule JA, Smith GF (1962) Akuamma alkaloids. Part I. Akuammine and χ-akuammigine. J Chem Soc 0:312–323
Julian PL (1931) PhD Dissertation, Vienna [quoted as ref 4 in (Julian and Pikl 1933)]
Julian PL (1933) Am Chem Soc meeting, Chicago [quoted as ref 104 in (Julian et al. 1952b)]
Julian PL (1952) Unpublished work [quoted as ref 378 in (Julian et al. 1952b)]
Julian PL, Magnani A (1949) Studies in the indole series. XII. Yohimbine (Part 3). A novel synthesis of the yohimbine ring structure. J Am Chem Soc 71:3207–3210
Julian PL, Magnani A (1952) Unpublished work [quoted as ref 102 in (Julian et al. 1952b)]
Julian PL, Passler W (1932) The thermal interconversion of mixed benzoins. J Am Chem Soc 54:4756
Julian PL, Passler W (1952) Unpublished work [quoted as ref 110 in (Julian et al. 1952b)]
Julian PL, Pikl J (1933) Studies in the indole series. I. The synthesis of alpha-benzylindoles. J Am Chem Soc 55:2105–2110
Julian PL, Pikl J (1935a) Studies in the indole series. III. On the synthesis of physostigmine. J Am Chem Soc 57:539–544
Julian PL, Pikl J (1935b) Studies in the indole series. IV. The synthesis of d,l-eserethole. 57:563–566
Julian PL, Pikl J (1935c) Studies in the indole series. V. The complete synthesis of physostigmine (eserine). J Am Chem Soc 57:755–757
Julian PL, Pikl J (1935d) The indole series, VII. The course of the Fischer reaction with ketones of the type RCH2CO CH3, ɑ-propyl- and ɑ-homoveratrylindole. Proc Indiana Acad Sci 45:145–150 [Chem Abs (1937) 31:10268]
Julian PL, Printy HC (1949) Studies in the indole series. XI. The reduction of certain oxindoles with lithium aluminum hydride. J Am Chem Soc 71:3206–3207
Julian PL, Printy HC (1952) Unpublished work [quoted as ref 354 in (Julian et al. 1952b)]
Julian PL, Printy HC (1953) Studies in the indole series. XIII. Oxindole -3- propionic acid. J Am Chem Soc 75:5301–5305
Julian PL, Pikl J, Boggess D (1934) Studies in the indole series. II. The alkylation of 1-methyl-3-formyloxindole and a synthesis of the basic ring structure of physostigmine. J Am Chem Soc 56:1797–1801
Julian PL, Pikl J, Wantz FE (1935) Studies in the indole series. VI. On the synthesis of oxytryptophan and further studies of 3-alkylation of oxindoles. J Am Chem Soc 57:2026–2029
Julian PL, Meyer EW, Krause NC (1940) Recovering sterols. U.S. 2, 218, 971, 22 Oct [Chem Abs (1941) 35; 10728] [and quoted in (Witkop 1998)]
Julian PL, Meyer EW, Magnani A, Cole W (1945) Studies in the indole series. IX. The reactions of ɑ-halogenated and ɑ-hydroxy ketones with arylamines. Part I. J Am Chem Soc 67:1203–1211
Julian PL, Magnani A, Pikl J, Karpel WJ (1948a) Studies in the indole series. VIII. Yohimbine (Part 1). The mechanism of dehydrogenation of yohimbine and related compounds. J Am Chem Soc 70:174–179
Julian PL, Karpel WI, Magnani A, Meyer EW (1948b) Studies in the indole series. X. Yohimbine (Part 2). The synthesis of yobyrine, yobyrone, and tetrahydroyobyrine. J Am Chem Soc 70:180–183
Julian PL, Karpel WJ, Magnani A, Meyer EW (1948c) The synthesis of ketoyobyrine. J Am Chem Soc 70:2834
Julian PL, Meyer EW, Ryden I (1949) 17ɑ-Hydroxysteroids. J Am Chem Soc 71:756
Julian PL, Meyer EW, Karpel WJ, Waller IR (1950) Sterols. XI. 17ɑ-hydroxy-11-desoxycorticosterone (Reichstein’s substance S). J Am Chem Soc 72:5145–5147
Julian PL, Meyer EW, Karpel WJ, Cole (1951) Sterols XII the partial synthesis of 4-pregnene -17ɑ, 20, 21-triol-3-ones and Reichstein’s substance. E J Am Chem Soc 73:1982–1985
Julian PL, Meyer EW, Printy HC (1952a) The chemistry of indoles. In: Elderfield RC (ed) Heterocyclic compounds, vol 3. Wiley/Chapman & Hall, limited, New York/London, chapter 1, pp 1–274
Julian PL et al (1952b) Unpublished work [quoted as ref 378 in (Julian et al. 1952b)]
Julian PL et al (1952c) J Am Chem Soc (in press) [quoted as refs 485 and 488 in (Julian et al. 1952b)]
Julian PL, Printy HC, Ketcham R, Doone R (1953) Studies in the indole series XIV. Oxindole -3-acetic acid. J Am Chem Soc 78:5305–5308
Julian PL, Cole JW, Meyer EW, Karpel WJ (1956a) Cortisone. U.S. 2, 752, 839, 26 June [Chem Abs (1957) 51:2081f-2083c] [and quoted in (Witkop 1998)]
Julian PL, Printy HC, Dailey EE (1956b) Studies in the indole series XV. Dioxindole-3-propionic acid. J Am Chem Soc 78:3501–3503
Julian PL, Dailey EE, Printy HC, Cohen HL, Hamashige S (1956c) Studies in the indole series. XVI. Oxindole-3-alanine and dioxindole-3-alanine. J Am Chem Soc 78:3503–3508
Kapil KS (1971) The carbazole alkaloids. In: Manske RHF (ed) The alkaloids chemistry and physiology, vol 13. Academic, New York/London, Chapter 6, pp 273–302
Kawabuchi M, Boyne AF, Deshpande SS, Cintra WM, Brossi A, Albuquerque EX (1988) Enantiomer (+) -physostigmine prevents organophosphate- induced subjunctional damage at the neuromuscular synapse by a mechanism not related to cholinesterase carbamylation. Synapse 2:139–147
Keimatsu S, Sagasawa S (1928) Synthesis of indole derivatives. I. Synthesis of physostigmol ethyl ether. J Pharm Soc Japan 48:348–355 [Chem Abs (1928) 22:32163]
Kermack WO, Robinson R (1922) LI. An explanation of the property of induced polarity of atoms and an interpretation of the theory of partial valencies on an electronic basis. J Chem Soc 121:427–440
Kikuchi T, Kadota S, Nakamura K, Nishi A, Taga T, Kaji OK, Tubaki K (1982) Dethio-tetra(methylthio)chetomin, a new antimicrobial metabolite of chaetomium globsum kinze ex fr. Structure and partial synthesis from chetomin. Chem Pharm Bull (Tokyo) 30(10):3846–3848
Kimura Y, Hamasaki T, Nakajima H, Isogai A (1982) Structure of aszonalenin, a new metabolite of aspergillus zonatus. Tetrahedron Lett 23(2):225–228
King FE, Robinson R (1932a) Experiments on the synthesis of physostigmine (eserine). Part V. A synthesis of dehydroesermetholomethine. J Chem Soc 326–336
King FE, Robinson R (1932b) Experiments on the synthesis of physostigmine (eserine). Part VI. A synthesis of dl-esermethole methopicrate. J Chem Soc 1433–1438
King FE, Robinson R (1933) Experiments on the synthesis of physostigmine (eserine). Part VII. J Chem Soc 270–273
King FE and Robinson R (1935) Experiments on the synthesis of physostigmine (eserine). Part XI. The later phases of the synthetical investigations. J Chem Soc 169:755–759
King FE, Robinson R, Suginome H (1933a) Experiments on the synthesis of physostigmine (eserine). Part VIII. J Chem Soc:1472–1475
King FE, Liguori M, Robinson R (1933b) Experiments on the synthesis of physostigmine (eserine). Part IX. An improvement in the synthesis of dl-eserethole. J Chem Soc 1475–1477
King FE, Liguori M, Robinson R (1934) Experiments on the synthesis of physostigmine (eserine). Part X. dl-Noresermethole and crystalline dl-eserethole. J Chem Soc 1416–1419
Kingscott PCR, Knight RSG (1914) Methods of quantitative organic analysis. Longmans, Green and Co, London, New York/Bombay/Calcutta/Madras, pp 93–104
Klein J (2007) Phenserine. Expert Opin Investig Drugs 16(7):1087–1097
Kobayashi T (1938) Über die konstitution des methyl-eseräthols und die optische spaltung von dl-eseräthol. Synthetische versuche über eserin V. Liebig’s Ann Chem 536:143–163
Kobayashi T (1939) Über die konstitution des methyl-eseräthols. 2. Synthetische versuche über eserin. VI. Liebig’s Ann Chem 539:213–218
Kolosov MN, Metroveli LI, Preobrazhenskii NA (1953) Synthetic investigations of indole derivatives, IV. The synthesis of esermethole, homoesermethole, and homoeseroline. J Gen Chem USSR 23:2413–2149; Zh Obsh Khim 23:2027–2034 [Chem Abs (1955) 49:3208i–3210c]
Kominck LA (1967) In: Gottlich D, Shaw PD (eds) Antibiotics, vol 2. Springer, Berlin, pp 231–239
Kupchan SM, Branfman AR, Snoden AT, Verma AK, jun DRE, Komoda Y, Hagao Y (1975) Novel maytansinoids. Naturally occurring and synthetic antileukemic esters of maytansinol. J Am Chem Soc 97:5294–5295
Kutney JP (1977) In: ApSimon (ed) The synthesis of indole alkaloids. Wiley-Inter- science, New York, pp 273–438
Lauter WM, Foote PA (1955) Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc Sci Ed 44:361–363
Laws I, Mantle PG (1985) Nigrifortine, a diketopiperazine metabolite of Penicillium nigricans. Phytochemistry 24(6):1395–1397
Lee TBK, Wong GSK (1990) High-performance liquid chromatographic enantioseparation of intermediates relating to the total synthesis of (-)-physostigmine. J Chromatogr 523:317–320
Lee TBK, Wong GSK (1991) Asymmetric alkylation of oxindoles: an approach to the total synthesis of (−)-physostigmine. J Org Chem 56:872–875. [see also (Grethe and Uskoković 1983)]
Lehninger AL (1970) Biochemistry. Worth Publishers Inc, New York
Longmore RB (1969) The absolute configurations of the alkaloids of Physostigma venenosum seeds. PhD Thesis, Victoria Manchester
Longmore RB, Robinson B (1966) Synthesis of l-physovenine: a new approach to the ring system. Chem Ind:1638–1639
Longmore RB, Robinson B (1967) Alkaloids of Physostigma venenosum. V. The synthesis of (±)-physovenine. Collect Czech Chem Commun 32:2184–2192. Presented at the symposium on chemistry and stereochemistry of steroid and indole alkaloids, held at Smolenice, Czechoslovakia on 14th–18th September 1965
Longmore RB, Robinson B (1969a) Absolute configuration of (-)–physostigmine and related alkaloids. Chem Ind 19:622–623
Longmore RB, Robinson B (1969b) The absolute configuration of the alkaloids of Physostigma venenosum seeds. J Pharm Pharmacol 21(1):118S–125S
Longmore RB, Robinson B (1973) Comments and further observations on the structure of eserine (physostigmine). Nat New Biol 246(155):239–240
Lovel FM (1953) Crystallographic data for certain alkaloids. III. Acta Cryst 6:869
Lucier JJ (1993) 1902 Nobel Laureate Emil Fischer 1852–1919. In: James LK (ed) (1993) 8–14 and bibliography quoted therein
Luo Y, Yu Q, Chrisey L, Brossi A (1990) Synthesis of (±)-physovenine and (±)-7-bromophysovenine from intermediates of the synthesis of physostigmines. Heterocycles 31(2):283–287
Luo W, Yu Q, Zhan M, Parrish D, Deschamps JR, Kulkarni SS, Holloway HW, Alley GM, Lahari DK, Brossi A, Greig NH (2005b) Novel anticholinesterases based on the molecular skeletons of furobenzofuran and methanobenzodioxepine. J Med Chem 48:986–994
Luo W, Yu Q, Kulkarni SS, Parrish DA, Holloway HW, Tweedie D, Shafferman A, Lahiri DK, Brossi A, Greig NH (2006) Inhibition of human acetyl˗ and butyrylcholinesterase by novel carbamates of (–)- and (+)-tetrahydrofurobenzofuran and methanobenzodioxepine. J Med Chem 49:2174–2185
Mahler HR, Cordes EH (1966) Biological chemistry. Harper and Row, New York
Mair AE, Miller JHMB (1984) Effect of sterilization by autoclaving on the stability of eye drops of pilocarpine hydrochloride and physostigmine sulphate. J Clin Hosp Pharm 9:217–224
Manohar H, Ramaseshan S (1961) The absolute configuration of echitamine iodide. Tetrahedron Lett (22): 814–816
Marino JP, Bogdan S, Kimura K (1992) The enantioselective synthesis of (-)-physostigmine via chiral sulfoxides. J Am Chem Soc 114:5566–5572
Marion L (1952) Alkaloids of Calabar bean. In: Manske RHF and Holmes HL (eds) The alkaloids, vol 2. Academic, New York, Chapter 11, pp 438–450, 494–497
Marki F, Robertson AV, Witkop B (1961) Dehydrobufotenine, a novel type of tricyclic serotonin metabolite from Bufo Marinus. J Am Chem Soc 83:3341–3342
Marques DIR (1957) Recientes progresos de la investigacion en el campo de los alcaloides de las papilionaceas. University of Santiago, Spain, 41 [quoted as ref 10 in (Robinson B 1964b) (see also Holmes 1879)]
Massart L, Vandendriessche L, Dufait R (1939) Autoxidation and enzyme oxidation of eseroline. Enzymologia 7:339–341 [Chem Abs (1940) 34: 63046]
Merck Index (1989) 11th edn, Index no 7357 [as quoted as ref 3 in (Yu et al. 1991) – nowhere in this Index is “urinary retention” even cited]
Merck Index (1996) 12th edn, Index no 7540
Merck Index (2001) 13th edn, Index no 1024(a), 3733(b), 5527(c), 6755(d) 6898(e), 7064(f), 7468(g), 7469(h), 7470(i), 7505(j), 8627 (k), 8882(l), 8893(m), 8909(n)
Miller CO (1954) Stable physostigmine solutions. US 2,678,899, 18 May [Chem Abs (1954) 48: 9632c]
Mitchell MO, Le Quesne PW (1990) Total synthesis of (±) – pseudophrynaminol. Tetrahedron Lett 31(19):2681–2684
Moroi SE, Lichter PR (1996) Ocular pharmacology. In: [Gilman et al. (eds) 1996], Chapter 65, pp 1619–1645
Muhtadi FJ, El-Hawary SS (1989) Analytical profile of physostigmine salicylate. In: Florey K (ed) Analytical profiles of drug substances, vol 18. Academic, San Diego/New York/Berkeley/Boston/London/Sydney/Tokyo/Toronto, pp 289–350
Murao S, Hayashi H (1986) Physostigmine and N8-norphysostigmine, insecticidal compounds from Streptomyces sp. Agric Biol Chem 50:523–524
Muthusubramanian P, Carlé JS, Christophersen C (1983) Marine alkaloids. 7. Synthesis of debromoflustramine B and related compounds. Acta Chem Scand B37:803–807
Naidu MB, Zaheer SH (1959) Stabilization of physostigmine sulphate. Indian 69,325 Oct 14 [Chem Abs (1962) 56:2519i]
Nakagawa M, Yoshikawa K, Hino T (1975) The photosensitized oxygenation of Nb-methyltryptamine. J Am Chem Soc 97:6496–6501
Nakano T, Martin A (1976) Studies on the alkaloids of Palicourea fendleri. Planta Med 30:186–188
Neuwinger HD (1996) Physostigma Venenosum Balfour. In: African ethnobotany poisons and drugs chemistry, pharmacology, toxicology, translated by Porter A and the author from the original German edition Afrikanische arzneipflanzen und jagdgifte. Chapman & Hall, London/Glasgow/Weinheim/New York/Tokyo/Melbourne/Madras, XVI, 699–707, colour plate -VI (this publication also includes a useful chapter upon literature concerning traditional medicine in Africa)
Newkome GR, Bhacca NS (1969) The absolute configuration of physostigmine (eserine). Application of the nuclear Overhauser effect. J Chem Soc Chem Commun:385
Niederl JB, Niederl V (1948) Micromethods of quantitative organic analysis, 2nd edn. Wiley/ Chapman & Hall Ltd, New York/London, pp 239–251
Node M, Hao XJ, Fuji K (1991) A chiral total synthesis of (-)-physostigmine. Chem Letters 20:57–60
Nwaji MN, Onyiriuka SO, Taylor DAH (1972) 6-(3-Methylbuta-1,3-dienyl)indole from Monodora tenuifolia. J Chem Soc Chem Commun:327
O’Donovan DG, Keogh MF (1966) The biosynthesis of folicanthine. J Chem Soc (C):1570–1572
Ohmomo S, Sato T, Utagawa T, Abe M (1975) Isolation of festuclavine and three new indole alkaloids, roquefortine A, B and C from the cultures of Pencillium roqueforti. Agric Biol Chem 39(6):1333–1334
Ohmomo S, Utagawa T, Abe M (1977) Identification of roquefortine C produced by Penicillium roqueforti. Agric Biol Chem 41(10):2097–2098
Ohmomo S, Oguma K, Ohashi T, Abe M (1978) Isolation of a new indole alkaloid, roquefortine D, from the cultures of Penicillium roqueforti. Agric Biol Chem 42(12):2387–2389
Onaka T (1971) One-step construction of 2,3,3a,8a-tetrahydrofuro[2,3-b]indole system: an application to physovenine synthesis. Tetrahedron Lett (46):4391–4392
Orekhov AP (1955) Chemistry of alkaloids, 2nd edn. Akademiia Nauk USSR, Moscow, 601 [quoted as ref 12 in (Robinson B 1964b)]
Pallavicini M, Valoti E, Villa L, Lianza F (1994) Synthesis of (-)- and (+)-esermethole via chemical resolution of 1,3-dimethyl-3-(2-aminoethyl)-5-methoxyoxindole. Tetrahedron Asymmetry 5:111–116
Pande PN, Gupta HL, Ameta SC, Bokadia MM (1979) Mass spectral behaviour of some deoxybisnoreserolines. Natl Acad Sci Lett (India) 2(5):178 [Chem Abs (1980) 92: 147011n]
Parry KP, Smith GF (1978) Quadrigemines -A and –B, two minor alkaloids of Hodgkinsonia frutescens F. Muell J Chem Soc (Perkin I) 1671–1682
Pauling P, Petcher TJ (1973) Crystal and molecular structure of eserine (physostigmine). J Chem Soc (Perkin II):1342–1345
Pavolini T, Gambarin F, Godenigo AS (1951) Fenossazine da 5,6-indolinchinoni. Gazz Chim Ital 81:527–532
Pei X, Bi S (1994) Reduction of oxindoles with sodium bis(2-methoxyethoxy) aluminum hydride, a novel reducing agent. Heterocycles 39(1):357–360
Pei X, Brossi A (1995) A facile preparation of (3aS)-1,3-dimethyl-3-cyanomethyl-5-ethoxyoxindole from Julian’s nitrile enriched in the (3aS)-enantiomer. Heterocycles 41:2823–2826
Pei X, Greig NH, Flippen-Anderson JL, Bi S, Brossi A (1994) 125 Total synthesis of racemate and optically active compounds related to physostigmine and ring-C heteroanalogues from 3-[3′-(dimethylamino) ethyl]-2-3-dihydro-5-methoxy -1,3- dimethyl-1H-indol-2-ol. Helv Chim Acta 77:1412–1422
Pei X, Greig NH, Bi S, Brossi A, Toome V (1995a) Inhibition of human acetylcholinesterase by unnatural (+)-(3aR)-N1-norphysostigmine and arylcarbamate analogues. Med Chem Res 5:265–270
Pei X, Greig NH, Bi S, Brossi A (1995b) Preparation and selective inhibition of human butyrylcholinesterase by N1-phenethylnorphysostigmine analogues. Med Chem Res 5:455–461
Pei X, Yu Q, Lu B, Greig NH, Brossi A (1996) Practical total syntheses of physostigmines and of phenserines: a synopsis. Heterocycles 42:229–236
Pei X, Greig NH, Brossi A (1998) Synthesis and biological evaluation of (±)-3a-phenyl congeners of physostigmine and phenserine. Heterocycles 49:437–444
Pelletier SW (1983) The nature and definition of an alkaloid. In: Pelletier SW (ed) Alkaloids chemical and biological perspectives. Wiley, New York/Chichester/Brisbane/Toronto/Singapore 1: chapter 1, pp 1–31
Perkin Jun WH, Robinson R (1906) Brazilin and haematoxylin. Part VIII. Proc Chem Soc 22:160–161
Perkin jun WH, Robinson R (1919) LXXIX. – Harmine and harmaline. Part III. J Chem Soc 115:933–967
Petcher TJ, Pauling P (1973) Cholinesterase inhibitors: structure of eserine. Nature 241:277
Petit A (1871) Sur une nouvelle matière colorante bloue dérivée de l’ésérine. CR. Acad Sci 62:569–570
Petit A, Polonovsky M (1893) Étude sur l’ésérine. Bull Soc Chim France 3rd. ser 9:1008–1015
Piloty O (1910) Synthese von pyrrolderivaten: pyrrole aus succinylobernsteinsäureester, pyrrole aus azinen. Ber 43:489–498
Plancher G (1898) Ueber die methylirung der indole. Ber 31:1488–1499
Polonovski M (1915) No 29 – Étude sur les alcaloides de la fève de Calabar (I). Esérine. Bull Soc Chim France 4th ser 17:235–244
Polonovski M (1916) No 8 – Étude sur les alcaloides de la fève de Calabar (V). Action de l’isocyanate de phényle: homologues phényliques de l’ésérine et de la génésérine. Bull Soc Chim France 4th ser 19:46–59
Polonovski M (1917) No 23. ˗ Etude sur les alcaloïdes de la fève de Calabar (VI). Constitution de la génésérine. Transformation de l’ésérine en génésérine. Bull Soc Chim France 4th ser 21:191–200
Polonovski M (1918) No 47. ˗ Etude sur les alcaloïdes de la fève de Calabar (VIII). Hydrogénation dans les series de l’ésérin, génésérine et ψ-génésérine. Bull Soc Chim France 4th ser 23:356–361
Polonovski M, Nitzberg C (1915a) No 30. ˗ Etude sur les alcaloïdes de la fève. de Calabar (II). La génésérine, nouval alcaloïde de la fève. Bull Soc Chim France 4th ser 17:244–256
Polonovski M, Nitzberg C (1915b) No 33. ˗ Etude sur les alcaloïdes de la fève de Calabar (III). Action de SO2 sur l’ésérine, la génésérine et leurs derives. Bull Soc Chim France 4th ser 17:290–297 (see also 289)
Polonovski M, Nitzberg C (1916) No 6 – Étude sur les alcaloides de la fève de Calabar (IV). Synthèse partielle de l’ésérine et de la génésérine. Bull Soc Chim France 4th ser 19: 27–37 [A correction to the authorship of this paper was noted in a subsequent publication (Polonovski 1916), footnote (1) of which states that “Dans le titre de la note (IV) (Bull. 1916, p. 27), le nom de M. Ch. Nitzberg s’est glissé par erreur. Le travail est de M. Polonovski seul”]
Polonovski M, Polonovski M (1918) No 46-Etude sur les alcaloides de la féve de Calabar (VII). Dégradation par iodométhylations successives des noyaux de l’ésérine et de la génésérine. Bull Soc Chim France, 4th Ser, 23:335–356 {on pages 215–217, in a preliminary communication of this work, it was stated that “M. Max POLONOVSKI, poursuivant ses recherches sur les alcaloïdes de la fève de Calabar présente une étude faite en collaboration avec M. Michel Polonovski, sur l’action de CH3I sur les bases de la série de l’ésérine ) et de la généserine” and it is interesting that footnote (1) on page 335 of this publication reports that “ La grande partie de ce travail fait en collaboration, date de 1913–1914, mais à cause des circonstances actuelles [presumably World War I from 1914 to 1918], la mise au point et la publication en ont été retardées jusqu’a ce jour”}
Polonovski M, Polonovski M (1923a) No 105 – Étude sur les alcaloides de la fève de Calabar (XI). Quelques hypotheses sur la constiution de l’ésérine. Bull Soc Chim France 4th ser 33:1117–1126
Polonovski M, Polonovski M (1923b) Sur la constitution de l’ésérine. CR Acad Sci Paris 176:1896–1898
Polonovski M, Polonovski M (1923c) Sur l’ésérétholmèthine et son alcoolate. CR Acad Sci Paris 177:127–129
Polonovski M, Polonovski M (1924a) No, 157. Étude sur les alcaloides de la fève de Calabar XIII. Formes tautomères de l’ésérines dérives nitrosés et benzoylés. Bull Soc Chim France 4th ser 35:1492–1522
Polonovski M, Polonovski M (1924b) Tautomèrie de l’ésérine. CR Acad Sci Paris 179:57–60
Polonovski M, Polonovski M (1924c) Dègradation des dérivés hydrés de l’ésérine. CR Acad Sci Paris 179:178–181
Polonovski M, Polonovski M (1924d) Dérivés nitrosés et benzoylés de l’ésérine. CR Acad Sci Paris 179:334–336
Polonovski M, Polonovski M (1924e) Sur les dérivés hydrés de l’ésérine. CR Acad Sci Paris 178:2078–2081
Polonovski M, Polonovski M (1925a) Sur l’oxyésérine et ses dérivés. CR Acad Sci Paris 180:73–76
Polonovski M, Polonovski M (1925b) No 60 - Étude sur les alcaloïdes de la fève de Calabar, XIV. Constitution de l’ésérine et des dérivés oxésériniques. Bull Soc Chim France 4th ser 37: 744–759
Polonovski M, Polonovski M (1925c) Sur les dérivés oxésériniques. CR Acad Sci Paris 180:1273–1275
Poobrasert O, Chal H, Pezzuto JM, Cordell GA (1996) Cytotoxic degradation product of physostigmine. J Nat Prod 59:1087–1089
Poobrasert O, Cordell GA, Bobzin SC (1997) Blue degradation products of rubreserine. J Nat Prod 60:578–580
Rahman A, Basha A (1983) Biosynthesis of indole alkaloids. Clarendon Press, Oxford, pp 159(a), 204(b), 205(c)
Rapoport H, Coxworth E (1959) Unpublished results [quoted as ref 49 in (Coxworth 1965)]
Rapoport H, Matteson DS, Gordon J, Coxworth E (1959a) Unpublished results [quoted as ref 48 in (Coxworth 1965)]
Rapoport H, Matteson DS, Gordon J, Coxworth E (1959b) Unpublished results [quoted as ref 91 “(private communication from E.C.)” in Abramovitch and Spenser 1964)]
Rege PD (2002) The introduction into oligomeric DNA of a number of abasic sites and their analogues and a new strategy for the synthesis of alkaloids of the Calabar bean family. PhD Dissertation, Part II: A new strategy for the synthesis of alkaloids of the Calabar bean family, State University of New York at Stony Brook
Rege PD, Johnson F (2003) Application of vicarious nucleophilic subsitution to the total synthesis of dl-physostigmine. J Org Chem 68:6133–6139
Remers WA (1972) Properties and reactions of indoles, isoindoles, and their hydrogenated derivatives In: Houlihan WJ (ed) indoles part one, vol 25. Wiley, New York/London/Sydney/Toronto, chapter I, pp 11–15
Riddell FG, Williams DAR, Hootelé C, Reid N (1970) Nitrogen inversion in tetrahydro-1,2-oxazines and the stereochemistry of geneserine. J Chem Soc (B):1739–1741
Ripperger H (1982) Chimonanthin aus Palicourea domingensis. Pharmazie 37:867
Robinson R (1917a) LXIII. A synthesis of tropinone. J Chem Soc 111:762–768
Robinson R (1917b) LXXV. A theory of the mechanism of the phytochemical synthesis of certain alkaloids. J Chem Soc 111:876–899
Robinson R (1925) Suggestion from [quoted in (Stedman and Barger 1925)]
Robinson R (1955) The structural relations of natural products. Being the first Weizmann memorial lectures December, 1953. The third lecture the indole group of the alkaloids. Clarendon Press, Oxford,pp 100–125, 138–141, (see, in particular, 103 and 104 for the proposed biogenesis of l-physostigmine)
Robinson B (1958) The synthesis and spectral investigation of:- dl-desoxybisnoreseroline, dl-iso-desoxynoreseroline, dl-desoxynoreseroline, dl-desoxyeseroline, and their derivatives. MSc Thesis, Victoria Manchester (see, in particular, pp 11–16)
Robinson B (1960) Some aspects of the chemistry of indoles and indole alkaloids. PhD Thesis, Victoria Manchester
Robinson B (1962) The autoxidation of 2-t-butyl-1,3,3-trimethylindolinol and 1,3,3-trimethyl-2-methyleneindoline (the Fischer base). Chem Ind:1291–1292
Robinson B (1963a) Alkaloids containing the Ph-N-C-N system. Chem Ind:218–227
Robinson B (1963b) The autoxidation of 2-hydroxy-1,3,3-trimethyl-2-t-butylindoline and 1,3,3-trimethyl-2-methyleneindoline. J Chem Soc:586–590
Robinson B (1964a) Alkaloids of Physostigma venenosum. Part I. The structure of physovenine. J Chem Soc:1503–1506
Robinson B (1964b) Alkaloids of Physostigma venenosum seeds. West Afr Pharm 6(5 and 6):92–96, 110–112
Robinson B (1964c) Unpublished observation [quoted as ref 14 in (Robinson B 1964b)]
Robinson B (1965a) Synthesis of dl-eseramine. Chem Ind 2:87–88
Robinson B (1965b) Alkaloids of Physostigma venenosum, part IV. The synthesis of (±) – eseramine. J Chem Soc 0:3336–3339
Robinson B (1965c) The structure of rubreserine, a decomposition product of physostigmine. J Pharm Pharmacol 17:89–91
Robinson B (1968) Alkaloids of the Calabar bean, In: Manske RHF (ed) The alkaloids chemistry and physiology, vol 10. Academic, New York/London, chapter 5, pp 383400
Robinson B (1969) The reduction of indoles and related compounds. Chem Rev 69:785–797
Robinson B (1971) Alkaloids of the Calabar bean. In: Manske RHF (ed) The alkaloids chemistry and physiology, vol 13. Academic, New York/London, Chapter 4, pp 213–226
Robinson R (1976) Memoirs of a minor prophet 70 years of organic chemistry. Elsevier, Amsterdam/Oxford/New York (see, in particular, p 17)
Robinson B (1982) The Fischer indole synthesis. Wiley, Chichester/New York/Brisbane/Toronto/Singapore, pp 1–923 and refs therein cited
Robinson B (1988a) The Calabar bean and its alkaloids: from magic to medicine. West African J Pharmacol Drug Res (J Quest – Africian de pharmacologie et de recherche sur les medicaments) 8:1–13 – a synopsis of presentations by the author to the 17th annual scientific conference concerning alkaloids in medicine of the West African Society for Pharmacology (Societe Quest-Africane de Pharmacologie), held at the University of Calabar, Nigeria, April 10th–13th, 1988
Robinson T (1988b) Alkaloids in bio-organic chemistry. Freeman, San Francisco and London, Chapter 18, p 168
Robinson B (2002) Syntheses of (-)-physostigmine, with particular emphasis upon the clarification of two enigmatic early synthetic approaches. Heterocycles, 57:1327–1352 [see also (Ault 2008)]
Robinson B, Hawkins DG (1985) Attempted Fischer indolisation of 2-(2-chloroethyl)cyclopentanone phenylhydrazone. Chem Ind:697–698
Robinson B, Moorcroft D (1970) Alkaloids of Physostigma venenosum, part IX. The absolute configuration of geneserine: an application of the nuclear Overhauser effect. J Chem Soc (C) 15:2077–2078
Robinson GM, Robinson R (1918) LIV – a new synthesis of tetraphenylpyrrole. J Chem Soc 113:639–645
Robinson GM, Robinson R (1924) C –the mechanism of E Fischer’s synthesis of indoles. Application of the method to the preparation of a pyrindole derivative. J Chem Soc 125:827–840
Robinson B, Robinson JB (1968) The anti-acetylcholinesterase activities of the alkaloids of Physostigma venenosum seeds. J Pharm Pharmacol 20(S1):213S–217S
Robinson B, Smith GF (1960) Indoles. Part IV. The reaction between 1,3-dimethylindole and mesityl oxide. J Chem Soc:4574–4578
Robinson R, Suginome H (1932a) Experiments on the synthesis of physostigmine (eserine). Part I. Some indolenine derivatives. J Chem Soc:298–304
Robinson R, Suginome H (1932b) Experiments on the synthesis of physostigmine (eserine). Part II. Synthesis of a base which is believed to be dl-noreserethole. J Chem Soc:304–317
Robinson R, Teuber HJ (1954) Calycanthine and calycanthidine. Chem Ind:783–784
Robinson B, Smith GF, Jackson AH, Shaw D, Frydman B, Deulofeu V (1961) Dehydrobufotenin. Proc Chem Soc:310–311
Robinson B, Rees JMH, Cox B (1987) (Victoria University of Manchester) new organic compounds having opioid properties. Int Pat Appl PCT/GB87/00457 30 June 1987 – see also Eur Pats Bull, Pub no EP0312539(A1)-1989-04-26
Robinson B, Rees JMH, Cox B (1988) (Victoria University of Manchester) Preparation of new echiboline derivatives having opioid properties. PCT Int. Appl. WO 88, 00,193 (Cl. CO7D487/08), 14 Jan 1988, GB appl. 86/16,089, 02 Jul 1986; 40pp. [Chem Abs (1988) 109:73726v]
Rodin FH (1947) Eserine: its history in the practice of ophthalmology. Am J Ophthal 30:19–28
Rosenmund P, Sadri E (1979) Beiträge zur chemie des indols, XII. Synthesen eserinähnlicher verbindungen, III; Neue an N-1 und N-8 substituierte derivate des desoxyeserolins sowie eine neue synthese des eserethols. Liebig’s Ann Chem (7):927–943
Rosenmund P, Sotiriou A (1964) Eine neuartige synthese des eserin – grundegerüstes. Angew Chem 76:787. [see also Angew Chem (Intern Edit) 3: 641]
Rosenmund P, Sotiriou A (1975) Beiträge zur chemie des indols, VI. Synthesen auf dem gebiet eserin-ähnlicher verbindugen, I. Eine neue synthese des eseringrundgerüstes. Chem Ber 108:208–214
Roth A, Kuballa B, Cabalion P, Anton R (1985) Preliminary study of the alkaloids of Psychotria forsteriana. Planta Med 51:289
Rubino FM, Zecca L (1991) Retrosynthetic fragmentation in the fast atom bombardment mass spectra of eserine and some related compounds. Org Mass Spec 26:961–966
Sainsbury M (1977) The synthesis of 6H-pyrido[4,3-b]carbazoles. Synthesis July: 437–448, 1977
Saito I, Matsuura T, Nakagawa M, Hino T (1977) Intermediates in photosensitized oxygenation of tryptophan derivatives. Acc Chem Res 10:746–352 and refs therein cited
Saleha S, Khan NH, Siddiqui AA, Kidwai MM (1978) Synthesis of indoles via cyclization of phenylhydrazones of keto compounds by formic acid. Indian J Chem 16B:1122–1124
Sallay SI (1967) The total synthesis of dl-ibogamine. J Am Chem Soc 89:6762–6763 [quoted in (Robinson B 1982)]
Saltzman M (1993) 1947 Nobel laureate Robert Robinson 1885–1875. In: James LK (ed) 306–314 and bibliography quoted therein
Salway AH (1911) CCXLII. Chemical examination of Calabar beans. J Chem Soc 99: 2148–2159 {see also Pharm J Pharmacist, 87: 719; Proc Chem Soc 27:273–274 [Chem Abs (1912) 6: 792]}
Salway AH (1912b) CI. Researches on the constitution of physostigmine. Part I. J Chem Soc 101:978–989 1589
Salway AH (1913) XLIII. – researches on the constitution of physostigmine. Part II. The synthesis of 3-dimethylaminoacetyl-2-methylindole and 2-α-dimethylamino-γ-hydroxypropylindole. J Chem Soc 103:351–361
Sammes PG (1975) Naturally occurring 2,5-dioxepiporazines and related compounds. Fortschr Chem Org Naturst 32:51–118
Sandoz Ltd (1966) Indoles. Neth Appl 6,608,066 (Cl C07d), Dec 19, 1966; U.S. Appl , June 17, 1965 [Chem Abs (1967) 67:82203p]
Savige WE (1975) New oxidation products of tryptophan. Aust J Chem 28:2275–2287
Savoury B, Turnbull JH (1985) Luminescence studies on physostigmine, rubreserine and adrenochrome. J Photochem 31:345–358
Saxton JE (1960) The alkaloids of Calabar bean. In: the indole alkaloids. In: Manske RHF (ed) The alkaloids, vol 7. Academic, New York/London, chapter 13, pp 146–147
Saxton JE (ed) (1983b) Indoles, part 4. The monoterpenoid indole alkaloids. In: Weissberger A and Taylor EC (eds) The chemistry of heterocyclic compunds, vol 25. Wiley, New York/Chichester/Brisbane/Toronto/Singapore, pp 1–886
Saxton JE, Bardsley WG, Smith GF (1962) The structure of calycanthidine. Proc Chem Soc 148
Schönenberger B, Brossi A (1986) 150. Fragmentation of optically active (1-phenylethyl)- and (1-naphthylethyl)ureas in refluxing alcohols: easy preparation of optically active amines of high optical purity. Helv Chim Acta 69:1486–1497
Schönenberger B, Brossi A, Clifford GC, Flippen-Anderson J (1986a) Preparation of optically active secondary amines by thermal decomposition of (methylbenzyl)urea analog – absolute configuration of (+)- and (-)- mecamylamine. Helv Chim Acta 69:283–287
Schönenberger B, Jacobson AE, Brossi A, Streaty R, Klee WR, Flippen-Anderson JL, Gilardi R (1986b) Comparison of (-)-eseroline with (+)-eseroline and dihydroseco analogues in antinociceptive assays: confirmation of rubreserine structure by X-ray analysis. J Med Chem 29:2268–2273
Schütte HR, Maier B (1965) Zur biosynthese der calycanthinalkaloide. Arch Pharm 298:459–461 [Chem Abs (1965) 63: 13708a]
Schwyzer J (1927) Die fabrikation der alkaloide. Springer, Berlin, 1–123 [quoted in ref 10 by (Henry 1949)] [Chem Abs (1928) 22: 1017] Die fabrikation pharmazeutischer und chemisch-technischer produkte. (Berlin, 1931), 338 {quoted in [Merck index 2001(h)]}
Scott AI, McCapra F and Hall ES (1964) Chimonanthine. A one-step synthesis and biosynthetic model. J Am Chem Soc 86:302–303 (see also: Total synthesis of alkaloids seems to follow biogenesis. Chem Eng News 24 Feb:40, 41)
Scott PM, Merrien M-A, Polonsky J (1976) Roquefortine and isofumigaclavine a, metabolites from Penicillium roqueforti. Experientia 32(2):140–142
Sherwood Taylor F (1948) A century of British chemistry. Longmans, Green and Co., London, New York, Toronto, for the British Council, 21, 31–33 and plates X and XI between 24 and 25
Shishido K, Fukumoto K (1988) Development of novel synthetic reaction using pericyclic reaction of o-quinodimethane and is application. Yuki Gosei Kagaku Kyokaishi (J Syn Org Chem Japan) 46(12):1179–1194 (in Japanese) and refs cited therein [Chem Abs (1989) 110:192274b]
Shishido K, Shitara E, Komatsu H, Hiroya K, Fukumoto K, Kametani T (1986b) Total synthesis of (±)-physovenine and (±)-physostigmine. An application of tandem electrocyclic -[3,3] sigmatropic reaction of benzocyclobutene. J Org Chem 51:3007–3011
Sourkes TL (1966) Nobel prize winners in medicine and physiology 1901–1965. Abelard-Schuman, London/New York/Toronto, pp 191–200(a), 278–290(b), 407–420(c)
Spande TF (1979) Hydroxyindoles, indole alcohols, and indolethiols. In: Houlihan WJ (ed) Indoles part three, vol 25. Wiley, New York/Cichester/Brisbane/Toronto, Chapter VIII, pp 7(a), 46–67(b), 84, 130 (c), 143–146(d), 167(e), 244(f), 252–253(g), 283(h), 441(i)
Spande TF, Wilchek M, Witkop B (1968) The reaction of derivatives of tryptophan, tryptamine, and other indoles with 2-hydroxy-5-nitrobenzyl bromide (Koshland’s reagent). J Am Chem Soc 90:3256–3258
Spande TF, Edwards MW, Pannell LK, Daly JW, Erspamer V, Melchiorri P (1988) Pseudophrynamine A: an unusual prenyl pyrrolo[2,3-b]indole ester from an Australian frog, Pseudophryne coriacea (myobatrachidae). J Org Chem 53:1222–1226
Späth E, Brunner O (1925) Zur konstitution des physostigmins. Ber 58:518–523
Späth E, Julian PL (1931) Neue corydalis alkaloide: d-tetrahydro-coptisin, d-canadin and hydro-hydrastinin. Ber 64B:1131–1137
Spiteller G, Spiteller-Friedmann M (1963) Schlüsselbruchstücke in den massenspektren von alkaloiden. Tetrahedron Lett 3:147–152
Spiteller G, Spiteller-Friedmann M (1964) Die massenspektrometrische untersuchung von naturstoffen und syntheseprodukten. Ind Chim Belge (4):357–371
Stedman E (1924) CLXXII. Physostigmine (eserine). Part II. The synthesis of physostigmol ethyl ether. J Chem Soc 125:1373–1376
Stedman E, Barger G (1925) XLII. Physostigmine (eserine). Part III. J Chem Soc 127:247–258
Stevens RV (1977) Alkaloid synthesis. In: ApSimon (ed) 1977, pp 439–553
Stillwell RN (1964) Total synthesis of ellipticine and 5-nor-ellipticine. PhD Thesis, Harvard [Dissert Abs (1964) 25:2769] [and quoted in (Sainsbury 1977), in which the relevant experimental details are also given, and as refs 4 and 16 in (Hewlins et al. 1984 and Gouyette et al. 1980, respectively), and in (Robinson B 1982)]
Stork G, Dolfini JE (1963) The total synthesis of dl-aspidospermine and of dl-quebrachamine. J Am Chem Soc 85:2872–2873
Straus F (1913) Zur kenntnis des physostigmin I. Liebig’s Ann Chem 401:350–376
Straus F (1914) Über physostigmin; [2. vorläufige mitteilung]. Liebig’s Ann Chem 406:332–341
Sugasawa S, Murayama M (1958a) Synthesis of dl-esermethole. Chem Pharm Bull (Tokyo) 6:194–200
Sugasawa S, Murayama M (1958b) Synthesis of homoesermethole. Chem Pharm Bull (Tokyo) 6:200–203
Sumpter WC, Miller FM (1954a) Calabar alkaloids. In : Sumpter and Miller FM (1954b), pp 203–208
Sumpter WC, Miller FM (1954b) Heterocyclic compounds with indole and carbazole systems. In: Weissberger A (ed) The chemistry of heterocyclic compounds, vol 8. Interscience Publishers, Inc, New York; Interscience Publishers Ltd, London, pp 1–307
Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I (1991) Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 253:872–879
Swallow W (1951) Preservation of eserine solutions. Pharm J 166:11
Takano S, Ogasawara K (1989) Alkaloids of the Calabar bean. In: Brossi (ed) The alkaloids chemistry and pharmacology, vol 36. Academic, San Diego/New York/Berkeley/Boston/London/Sydney/Tokyo/Toronto. Chapter 5, pp 225–251
Takano S, Moriya M, Iwabuchi Y, Ogasawara K (1990a) A chiral route to both enantiomers of physostigmine and the first synthesis of (−)-norphysostigmine. Chem Letters 19:109–112
Takano S, Sato T, Inomata K, Ogasawara K (1990b) An enantio controlled route to unnatural (+)-physostigmine. Heterocycles 31:411–414
Takano S, Moriya M, Ogasawara K (1991) Enantiocontrolled total synthesis of (−)-physovenine and (−)-physostigmine. J Org Chem 56:5982–5984
Takase S, Iwami M, Ando T, Okamoto M, Yoshida K, Horiai H, Kohsata M, Aoki H, Imanaka H (1984) Amauromine, a new vasodilator – taxonomy, isolation and characterization. J Antibiot 37(11):1320–1323
Takase S, Itoh Y, Uchida I, Tanaka H, Aoki H (1985a) Total synthesis of amauromine. Tetrahedron Lett 26(7):847–850
Takase S, Kawai Y, Uchida I, Tanaka H, Aoki H (1985b) Structure of amauromine, a new hypotensive vasodilator produced by amauroascus SP. Tetrahedron 41(15):3037–3048
Takase S, Uchida I, Tanaka H, Aoki H (1986a) Synthesis of debromo-8, 8a-dihydroflustramine C1, a model synthesis toward amauromine. Tetrahedron 42(21):5879–5886
Takase S, Itoh Y, Uchida I, Tanaka H, Aoki H (1986b) Total synthesis of amauromine. Tetrahedron 42(21):5887–5894
Tanganyika Rep Govt Chem (1952) [quoted as ref T59 in (Watt and Breyer-Brandwijk 1962)]
Taylor WI (1966) Indole alkaloids. An introduction to the enamine chemistry of natural products, vol 29. Pergamon Press, Oxford/London/Edinburgh/New York/Toronto/Paris/Braunschweig, pp 29, 30, 33–36, 145
Taylor P (1996) Anticholinesterase agents. In: Gilman et al. (eds) 1996, Chapter 8, pp 161–176
The Percy Julian biography project (2002) (preliminary treatment, 1st February (WGBH Educational Foundation, 125 Wester Ave, Boston, MA 02134) (script made available by Dr Bernhard Witkop on 11th September 2002); Percy Julian – forgotten genius; NOVA 492. http://www.pbs.org/wgbh/nova/julian/ [quoted as ref 1 in (Ault 2008)]. This represented a two-hour public television biography of a remarkable black scientist, entrepreneur and civil rights advocate
Tokuyama T, Daly JW (1983) Steroidal alkaloids (batrachotoxins and 4β-hydroxybatrachotoxins), “indole alkaloids” (calycanthine and chimonanthine) and a piperidinyldipyridine alkaloid (noranabasamine) in skin extracts from the Colombian poison-dart frog phyllobates terribilis (dendrobatidae). Tetrahedron 39:41–47
Travecedo EF, Stenberg VI (1970) Nitrogen photochemistry VIII. Photodecarbamation, a new reaction of physostigmine. Tetrahedron Lett 11:4539–4542
Triggle DJ, Mitchell JM, Filler R (1998) The pharmocolgy of physostigmine. CNS Drug Rev 4(2):87–136. and refs therein cited
Troxler F (1972) Indoles carrying basic nitrogen functions. In: Houliham WJ (ed) Indoles. Part II of The chemistry of heterocyclic compounds, vol 25. Wiley – Interscience, a division of Wiley, New York/London/Sydney/Toronto, chapter 6, p 227
Tsukamoto S, Hirota H, Kato H, Fusetani N (1993) Urochordamines a and B: larval settlement / metamorphosis- promoting, pteridine-containing physostigmine alkaloids from the tunicate Ciona savigni. Tetrahedron Lett 34(30):4819–4822
Udenfriend S, Titus E, Weissbach H, Peterson RE (1956) Biogenesis and metabolism of 5-hydroxyindole compounds. J Biol Chem 291:335–344
Vée A, Leven M (1864) De l’alcaloide de la fève du Calabar et expériences physiologiques avec ce même alcaloide. CR Soc Biol 160–172 [quoted as ref 44b in (Holmstedt 1972)]; Gazette Médicale, Paris 782–783[quoted in (Neuwinger 1996)]
Vée A, Leven M (1865) Recherches chimiques et physiologiques sur la fève du Calabar. Thèse, Paris [quoted in Jahresber Chem (1865) 456–457, Hesse O (1867), Neuwinger (1996) and as ref 44a in Holmstedt (1972)] – see also (Vée and Leven 1864)
Vincent D and Maugein A (1942a) Biochemical determination of eserine by measuring the inhibition of cholinesterase (application in medicine and pharmacy). Bull Sci Pharmacol 49:141–145 [Chem Abs (1944) 38:36807]
Vincent D, Maugein A (1942b) Biochemical determination of eserine by its anticholinesterase action. Its use in materia medica and pharmacy. Bull Sci Pharmacol 49:165–167 [Chem Abs (1944) 38:55195]
Vleggaar R, Wessals PL (1980) Stereochemistry of the dehydrogenation of (2S)-histidine in the biosynthesis of roquefortine and oxaline. J Chem Soc Chem Commun:160–162
Wani MC, Taylor HL, Wall ME (1973) Plant antitumour agents: colubrinol acetate and colubrinol, antileukaemic ansa macrolides from Colubrina texensis. J Chem Soc Chem Commun 390
Watt JM, Breyer-Brandwijk MG (1962) The medicinal and poisonous plants of southern and eastern Africa. E & S Livingstone Ltd., Edinburgh/London, pp 1–1457
Weber HP (1972) The molecular structure and absolute configuration of chaetocin. Acta Cryst B28:2945–2951
Wenkert E (1959) Alkaloid biosynthesis. Experientia 15(5):165–204 and refs quoted therein
Wieland H, Konz W, Mittasch H (1934) Die konstitution von bufotenin und bufotenidin. Uber kröten-gifstoffe VII. Liebig’s Ann Chem 513:1–25
Wildman WC (1970) Colchicine. In: Pelletier SW (ed) Chemistry of the alkaloids. Van Nostrand Reinhold Company, New York/Cincinnati/Toronto/London/Mebourne, Chapter 8, p 200
Witkop B (1950) The structure of quinamine. J Am Chem Soc 72:2311–2312
Witkop B (1956) Imine-enamine systems and the mechanism of their oxidation. J Am Chem Soc 78:2873–2882
Witkop B (1978) The story behind the story. The story of the oxidation of tryptophan as documented in the letters of Percy L Julian. Unpublished manuscript submitted to J Chem Educ in 1978 [cf Witkop (1983)] Witkop (2002) personal communication [the ample correspondence between Julian and Witkop which accumulated over a time span of almost 60 years is now in the archives of the Chemical Heritage Foundation in Philadelphia (Witkop 1998)] – see, also, Julian PL unpublished work [quoted as ref 378 in (Julian et al. 1952b)]
Witkop B (1980) Percy Lavon Julian (1899–1975). A biographical memoir. In: Biographical memoirs. The National Academy Press, Washington DC 52:223 [quoted in (Anon 1999) and as ref 19 in (Witkop 1983)]
Witkop B (1983) Forty years of trypto-fun. Heterocycles 20:2059–2075
Witkop B (1998) From the ordeal bean (physostigma venenosum) to the ordeal of Alzheimer’s disease - some of the legacy of Percy Lavon Julian (1899–1975). Heterocycles 49:9–27
Witkop B, Hill RK (1955) The synthesis of 2-carboxydeoxyeserolines via β-methyl-Ψ-tryptophan. J Am Chem Soc 77:6592–6595
Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K (1954) The total synthesis of strychnine. J Am Chem Soc 76:4749–4751
Woodward RB, Yang NC, Katz TJ, Clark VM, Harley-Mason J, Ingleby RFJ, Sheppard N (1960) Calycanthine: the structure of the alkaloid and its degradation product, calycanine. Proc Chem Soc:76–78
Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K (1963) The total synthesis of strychnine. Tetrahedron 19:247–288 [quoted in (Robinson B 1982)]
Yamada S, Hino T, Ogawa K (1963) The reductive cyclisation of 1-methyl-3-aminoalkyloxindoles with lithium aluminium hydride. Chem Pharm Bull (Tokyo) 11:674–678
Yamada T, Iwamoto C, Yamagaki N, Yamanouchi T, Minoura K, Yamori T, Uehara Y, Andoh T, Umemura K, Numata A (2002) Leptosins M-N1, cytotoxic metabolites from a Leptosphaeria species separated from a marine alga. Structure determination and biological activites. Tetrahedron 58:479–487
Yu Q, Brossi A (1988) Practical synthesis of unnatural (+)-physostigmine and carbamate analogues. Heterocycles 27:745–750
Yu Q, Schönenberger B, Brossi A (1987) Reactions of (−)-physostigmine and (−)-N-methylphysostigmine in refluxing butanol and at high temperature: facile preparation of (−)-eseroline. Heterocycles 26:1271–1275
Yu Q, Atack JR, Rapoport SI, Brossi A (1988a) Synthesis and anticholinesterase activity of (-)-N1-norphysostigmine, (-)-eseramine, and other N(1)-substituted analogues of (-)-physostigmine. J Med Chem 31:2297–2300
Yu Q, Atack JR, Rapoport SI, Brossi A (1988b) Carbamate analogues of (−)-physostigmine: in vitro inhibition of acetyl- and butyrylcholinesterase. FEBS Lett 234(1):127–130
Yu Q, Yeh HJC, Brossi A, Flippen-Anderson JL (1989) Geneserine and geneseroline revisited: acid-base catalyzed equilibria of hexahydropyrrolo-[2,3-b]-indole N-oxides with hexahydro-1,2-oxazino-[5,6-b]-indoles. J Nat Prod 52:332–336
Yu Q, Liu C, Brzostowaka M, Chrisey L, Brossi A, Greig NH, Atack JR, Soncrant TT, Rapoport SI, Radunz H-E (1991) Physovenines: efficient synthesis of (−)- and (+)-physovenine and synthesis of carbamate analogues of (−)-physovenine. Anticholinesterase activity and analgesic properties of optically active physovenines. Helv Chim Acta 74:761–766
Yu Q, Luo W, Li Y, Brossi A (1993) Progress towards a practical total synthesis of Calabar alkaloids: total synthesis of (−)-esermethole and (−)-physovenol methyl ether from (3S)-1,3-dimethyl-3-carboxymethyl-5-methoxyindole. Heterocycles 36:1279–1285
Yu Q, Lu B, Pei X (1994) Total synthesis of racemic physostigmine, physovenine and its sulphur analogue by the oxindole-5-O-tetrahydropyranyl ether route. Heterocycles 39(2):519–525
Yu Q, Zhu X, Holloway HW, Whittaker NF, Brossi A, Greig NH (2002) Anticholinesterase activity of compounds related to geneserine tantomers N-oxides and 1,2-oxides. J Med Chem 45:3684–3691
Zeisel Dr S (1895) Über ein verfahren zum quantitativen nachweise von methoxyl. Monatsh Chem 6:989–996
Zhao B, Moochhala SM, Tham S (2004) Biologically active components of Physostigma venenosum. J Chrom B 812:183–192
Zhigulev KK, Khmel’nitskii RA, Grandberg II, Vysotskii VI (1972) Indoles XXVII. Mass spectrometry of compounds with eserine and homoeserine skeletons. Chem Het Comps 8:962–965
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature B.V.
About this chapter

Cite this chapter
Robinson, B. (2023). l-Physostigmine (Eserine). In: The Calabar Bean and its Alkaloids. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1191-1_2
Download citation
DOI: https://doi.org/10.1007/978-94-024-1191-1_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-1190-4
Online ISBN: 978-94-024-1191-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)