
New Compounds from the Roots of Corsican Calicotome Villosa (Poir.) Link.: Two Pterocarpans and a Dihydrobenzofuran

 ,
,
Abstract
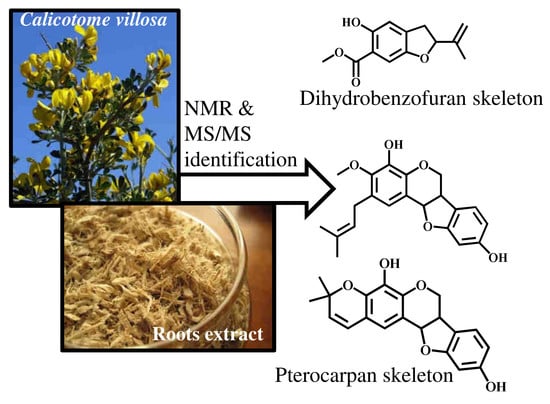
:
1. Introduction
2. Results and Discussion
- Compound 1, the major component of fraction F2, was isolated in pure form by repetitive CC and its structure elucidated using a combination of spectroscopic techniques including 2D NMR “INADEQUATE” (Incredible Natural Abundance DoublE QUAntum Transfer Experiment) sequence.
- As observed on their 13C NMR spectra, the sub-fractions F3.B.3 and F3.B.7 (see experimental) contained only the unidentified compounds 2 and 3 with the relative ratios 3:1 and 1:4, respectively, according to the relative intensities of signals. Although we did not succeed in isolating 2 and 3 in a pure form, the full set of 1D and 2D NMR experiments was conducted on the sub-fractions and compounds 2 and 3 were unambiguously identified.
2.1. Determination of 2-(1-methylethenyl)-5-hydroxy-6-carbomethoxy-2,3-dihydrobenzofuran (1)
2.2. Determination of 4,9-dihydroxy-3-methoxy-2-dimethylallylpterocarpan (2)
2.3. Determination of 4,9-dihydroxy-3′,3′-dimethyl-2,3-pyranopterocarpan (3)
3. Materials and Methods
3.1. Plant Material
3.2. Extraction and Isolation
3.3. NMR Spectroscopy
3.4. Mass Spectrometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jeanmonod, D.; Gamisans, J. Flora Corsica, 2nd ed.; Société Botanique du Centre-Ouest: Jarnac, France, 2013; p. 452. [Google Scholar]
- Loy, G.; Cottiglia, F.; Garau, D.; Deidda, D.; Pompei, R.; Bonsignore, L. Chemical composition and cytotoxic and antimicrobial activity of Calycotome villosa (Poiret) Link leaves. Il Farm. 2001, 56, 433–436. [Google Scholar] [CrossRef]
- El Antri, A.; Messouri, I.; Chendid Tlemçani, R.; Bouktaib, M.; El Alami, R.; El Bali, B.; Lachkar, M. Flavone glycosides from Calycotome villosa subsp. intermedia. Molecules 2004, 9, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Dessí, M.A.; Deiana, M.; Rosa, A.; Piredda, M.; Cottiglia, F.; Bonsignore, L.; Deidda, D.; Pompei, R.; Corongiu, F.P. Antioxidant activity of extracts from plants growing in Sardinia. Phytother. Res. 2001, 15, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, I.; Allali, H.; Bechlaghem, K.; Fekih, N.; Muselli, A.; Djabou, N.; El Amine Dib, M.; Tabti, B.; Halla, N.; Costa, J. Assessment of in vitro antimicrobial potency and free radical scavenging capacity of the essential oil and ethanol extract of Calycotome villosa subsp. intermedia growing in Algeria. Asian Pac. J. Trop. Dis. 2014, 4, 356–362. [Google Scholar] [CrossRef]
- Elkhamlichi, A.; El Hajaji, H.; Faraj, H.; Alami, A.; El Bali, B.; Lachkar, M. Phytochemical screening and evaluation of antioxidant and antibacterial activities of seeds and pods extracts of Calycotome villosa subsp. Intermedia. J. Appl. Pharm. Sci. 2017, 7, 192–198. [Google Scholar]
- Alhage, J.; Elbitar, H.; Taha, S.; Guegan, J.-P.; Dassouki, Z.; Vives, T.; Benvegnu, T. Isolation of bioactive compounds from Calicotome villosa stems. Molecules 2018, 23, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistelli, L.; Fiumi, C.; Morelli, I.; Giachi, I. Flavonoids from Calicotome villosa. Fitoterapia 2003, 74, 417–419. [Google Scholar] [CrossRef]
- El Antri, A.; Messouri, I.; Bouktaib, M.; El Alami, R.; Bolte, M.; El Bali, B.; Lachkar, M. Isolation and X-ray crystal structure of a new isoquinoline-N-oxide alkaloid from Calycotome villosa subspintermedia. Fitoterapia 2004, 75, 774–778. [Google Scholar] [CrossRef]
- El Antri, A.; Messouri, I.; Bouktaib, M.; El Alami, R.; Bolte, M.; El Bali, B.; Lachkar, M. Isolation and X-ray crystal structure of tetrahydroisoquinoline alkaloids from Calycotome villosa subsp. intermedia. Molecules 2004, 9, 650–657. [Google Scholar] [CrossRef] [Green Version]
- Cherkaoui-Tangi, K.; Lachkar, M.; Wibo, M.; Morel, N.; Gilani, A.H.; Lyoussi, B. Pharmacological studies on hypotensive, diuretic and vasodilator activities of chrysin glucoside from Calycotome villosa in rats. Phytother. Res. 2008, 22, 356–361. [Google Scholar] [CrossRef]
- Elkhamlichi, A.; El Antri, A.; El Hajaji, H.; El Bali, B.; Oulyadi, H.; Lachkar, M. Phytochemical constituents from the seeds of Calycotome villosa subsp. intermedia. Arab. J. Chem. 2017, 10, S3580–S3583. [Google Scholar] [CrossRef] [Green Version]
- Guaâdaoui, A.; El-Alami, I.; Abid, M.; Boukhatem, N.; Lechkar, M.; Hamal, A. Contribution to botanical, phyto-ecological and phytochemical studies of Calicotome villosa (Poiret) Link subsp. intermedia: A phylogenetic approach from Moroccan species. IJGHC 2016, 5, 93–111. [Google Scholar]
- Obara, Y.; Matsubara, H.; Munakata, K. Isolation and identification of tubaic acid and β-tubaic acid from Derris roots. Agric. Biol. Chem. 1976, 40, 1245–1246. [Google Scholar]
- Fotso, G.W.; Maher, F.A.; Ngnintedo, D.; Ango, P.Y.; Kapche, D.G.F.W.; Ngameni, B.; Ngwenya, B.; Yeboah, S.O.; Ngadjui, B.T.; Andrae-Marobela, K. Three new isoflavonoids with antioxidant properties from Ptycholobium contortum (Leguminosae). Phytochem. Lett. 2015, 14, 254–259. [Google Scholar] [CrossRef]
- Yoon, J.S.; Sung, S.H.; Park, J.H.; Kim, Y.C. Flavonoids from Spatholobus suberectus. Arch. Pharm. Res. 2004, 27, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Ingham, J.L.; Tahara, S.; Dziedzic, S.Z. Coumestans from the roots of Pueraria mirifica. Z. Naturforsch. 1988, 43, 5–10. [Google Scholar] [CrossRef]
- Tahara, S.; Ingham, J.L.; Dziedzic, S.Z. Structure elucidation of kwakhurin, a new prenylated isoflavone from Pueraria mirifica roots. Z. Naturforsch. 1987, 42, 510–518. [Google Scholar] [CrossRef]
- Mori-Hongo, M.; Takimoto, H.; Katagiri, T.; Kimura, M.; Ikeda, Y.; Miyase, T. Melanin synthesis inhibitors from Lespedeza floribunda. J. Nat. Prod. 2009, 72, 194–203. [Google Scholar] [CrossRef]
- Ingham, J.; Tahara, S. Isoneorautenol and other pterocarpan phytoalexins from Calopogonium mucunoides. Z. Naturforsch. 1985, 40, 482–489. [Google Scholar] [CrossRef]
- Jiménez-Gonzàlez, L.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Pterocarpans: Interesting natural products with antifungal activity and other biological properties. Phytochem. Rev. 2008, 7, 125–154. [Google Scholar] [CrossRef]
- Goel, A.; Kumar, A.; Raghuvanshi, A. Synthesis, stereochemistry, structural classification and chemical reactivity of natural pterocarpans. Chem. Rev. 2013, 113, 1614–1640. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.; Vincken, J.P.; Bohin, M.C.; Kuijpers, T.F.M.; Verbruggen, M.A.; Gruppen, H. Identification of prenylated pterocarpans and other isoflavonoids in Rhizopus spp. elicited soya bean seedlings by electrospray ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 55–65. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| N°C | δ13C (ppm) | DEPT | δ1H (ppm) | Multiplicity (J (Hz)) | HMBC (H→C) | COSY | NOESY |
| 2 | 85.52 | CH | 5.13 | t (8.0) | 11 | 3a; 3b | 3a; 3b; 12 |
| 3 | 35.27 | CH2 | 3.02 (a) 3.33 (b) | ddd (16.7; 8.0; 1.2) ddd (16.7; 8.0; 1.2) | 2; 4; 5; 8; 9; 10 | 2; 3b; 4 2; 3a; 4 | 2; 4; 12 2; 4; 12 |
| 4 | 113.79 | CH | 6.80 | br s | 3; 5; 6; 7; 8; 9; 13 | 3a; 3b | 3a; 3b; 5-OH |
| 5 | 156.86 | C | - | - | - | - | - |
| 6 | 110.48 | C | - | - | - | - | - |
| 7 | 107.40 | CH | 7.16 | s | 4; 5; 6; 8; 9; 13 | - | - |
| 8 | 152.34 | C | - | - | - | - | - |
| 9 | 136.55 | C | - | - | - | - | - |
| 10 | 143.67 | C | - | - | - | - | - |
| 11 | 112.23 | CH2 | 4.91(a) 5.07(b) | m m | 2; 10; 12 2; 3; 10; 12 | 12 | 11b; 12 11a; 12 |
| 12 | 17.17 | CH3 | 1.75 | br s | 2; 10; 11 | 11a; 11b | 2; 3a; 3b; 11a; 11b |
| 13 | 170.63 | C | - | - | - | - | - |
| 14 | 52.21 | CH3 | 3.91 | s | 13 | - | - |
| 5-OH | - | - | 10.55 | s | 4; 5; 6; 9 | - | 4 |
 | |||||||
|---|---|---|---|---|---|---|---|
| N°C | δ13C (ppm) | DEPT | δ1H (ppm) | Multiplicity (J (Hz)) | HMBC (H→C) | COSY | NOESY |
| 1 | 121.00 | CH | 6.89 | br s | 1′; 3; 4; 4a; 11a | - | 1′; 11a |
| 2 | 128.72 | C | - | - | - | - | |
| 3 | 145.52 | C | - | - | - | - | |
| 4 | 137.72 | C | - | - | - | - | |
| 4a | 142.01 | C | - | - | - | - | |
| 6 | 67.04 | CH2 | 4.32 (x) 3.62 (y) | dd (10.9;4.8) t (10.9) | 4a; 6a; 6b; 11a | 6y; 6a 6x; | 6y 6x |
| 6a | 39.70 | CH | 3.57 | m | 6; 6b; 7; 10a | 11a; 6x | 7; 11a |
| 6b | 118.99 | C | - | - | - | - | |
| 7 | 125.01 | CH | 7.08 | d (8.6) | 6a; 9; 10; 10a | 8 | 6a; 8 |
| 8 | 107.75 | CH | 6.37 | m | 10; 10a | 7 | 7 |
| 9 | 157.02 | C | - | - | - | - | |
| 10 | 98.48 | CH | 6.39 | s | 6b;8;9 | - | 9-OH |
| 10a | 160.70 | C | - | - | - | - | |
| 11a | 78.44 | CH | 5.50 | d (6.6) | 1; 4a; 6; 6a; 11b | 6a | 1; 6a |
| 11b | 115.52 | C | - | - | - | - | |
| 1′ | 28.15 | CH2 | 3.33 | m | 1; 2; 2′; 3; 3′ | 2′; 4′; 5′ | 1; 2′; -OCH3 |
| 2′ | 122.81 | CH | 5.29 | m | 4′; 5′ | 1′; 4′; 5′ | 1′ |
| 3′ | 132.46 | C | - | - | - | - | |
| 4′ | 17.85 | CH3 | 1.74 | br s | 2′; 3′; 5′ | 1′; 2′ | |
| 5′ | 25.83 | CH3 | 1.74 | br s | 2′; 3′; 4′ | 1′; 2′ | |
| -OCH3 | 60.62 | - | 3.86 | s | 3 | - | 1′ |
| 4-OH | - | - | 5.47 | s | 3; 4; 4a | - | - |
| 9-OH | - | - | 4.95 | s | 8; 9; 10 | - | 10 |
 | |||||||
|---|---|---|---|---|---|---|---|
| N°C | δ13C (ppm) | DEPT | δ1H (ppm) | Multiplicity (J (Hz)) | HMBC (H→C) | COSY | NOESY |
| 1 | 118.41 | CH | 6.77 | s | 1′; 2; 3; 4; 4a; 11a | - | 1′; 11a |
| 2 | 116.12 | C | - | - | - | - | |
| 3 | 140.30 | C | - | - | - | - | |
| 4 | 133.01 | C | - | - | - | - | |
| 4a | 143.73 | C | - | - | - | - | |
| 6 | 66.92 | CH2 | 4.34 (x) 3.63 (y) | dd (10.8; 5.0) t (10.8) | 4a; 6a; 6b; 11a | 6y; 6a 6x | 6y; 6a 6x; |
| 6a | 39.53 | CH | 3.53 | m | 6; 6b; 7; 10a; 11a | 11a; 6x | 6x; 11a |
| 6b | 119.00 | C | - | - | - | - | - |
| 7 | 125.04 | CH | 7.08 | d (8.3) | 6; 6a; 6b; 9; 10; 10a | 8 | 8 |
| 8 | 107.77 | CH | 6.37 | m | 6b | 7 | 7 |
| 9 | 157.07 | C | - | - | - | - | |
| 10 | 98.44 | CH | 6.38 | s | 6b; 8; 9; 10a | - | |
| 10a | 160.63 | C | - | - | - | - | |
| 11a | 78.57 | CH | 5.48 | d (6.5) | 1; 2; 4a; 6; 6a; 11b | 6a | 1; 6a |
| 11b | 112.65 | C | - | - | - | - | |
| 1′ | 121.79 | CH | 6.31 | d (9.8) | 1; 2; 3; 3′; 4; 5′ | 2′ | 1; 2′ |
| 2′ | 129.39 | CH | 5.57 | d (9.8) | 2; 3; 3′; 4′ | 1′ | 1′; 4′; 5′ |
| 3′ | 77.31 | C | - | - | - | - | - |
| 4′ * | 28.16 | CH3 | 1.48 | s | 1′; 2′; 3; 3′; 5′ | 2′ | |
| 5′ * | 27.90 | CH3 | 1.44 | s | 1′; 2′; 3′; 4′ | 2′ | |
| 4-OH | - | - | 5.38 #,$ | s | - | - | - |
| 9-OH | - | - | 5.28 #,$ | m | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palu, D.S.; Paoli, M.; Casabianca, H.; Casanova, J.; Bighelli, A. New Compounds from the Roots of Corsican Calicotome Villosa (Poir.) Link.: Two Pterocarpans and a Dihydrobenzofuran. Molecules 2020, 25, 3467. https://doi.org/10.3390/molecules25153467
Palu DS, Paoli M, Casabianca H, Casanova J, Bighelli A. New Compounds from the Roots of Corsican Calicotome Villosa (Poir.) Link.: Two Pterocarpans and a Dihydrobenzofuran. Molecules. 2020; 25(15):3467. https://doi.org/10.3390/molecules25153467
Chicago/Turabian StylePalu, Doreen Stacy, Mathieu Paoli, Hervé Casabianca, Joseph Casanova, and Ange Bighelli. 2020. "New Compounds from the Roots of Corsican Calicotome Villosa (Poir.) Link.: Two Pterocarpans and a Dihydrobenzofuran" Molecules 25, no. 15: 3467. https://doi.org/10.3390/molecules25153467