
Recent Advances in the Synthesis of β-Carboline Alkaloids




Egis Pharmaceuticals Plc., Directorate of Drug Substance Development, P.O. Box 100, H-1475 Budapest, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(3), 663; https://doi.org/10.3390/molecules26030663
Submission received: 13 November 2020
/
Revised: 16 January 2021
/
Accepted: 21 January 2021
/
Published: 27 January 2021
(This article belongs to the Special Issue Nitrogen-Containing Heterocycles as Significant Molecular Scaffolds for Medicinal and Other Applications)
Abstract
:β-Carboline alkaloids are a remarkable family of natural and synthetic indole-containing heterocyclic compounds and they are widely distributed in nature. Recently, these alkaloids have been in the focus of interest, thanks to their diverse biological activities. Their pharmacological activity makes them desirable as sedative, anxiolytic, hypnotic, anticonvulsant, antitumor, antiviral, antiparasitic or antimicrobial drug candidates. The growing potential inherent in them encourages many researchers to address the challenges of the synthesis of natural products containing complex β-carboline frameworks. In this review, we describe the recent developments in the synthesis of β-carboline alkaloids and closely related derivatives through selected examples from the last 5 years. The focus is on the key steps with improved procedures and synthetic approaches. Furthermore the pharmacological potential of the alkaloids is also highlighted.
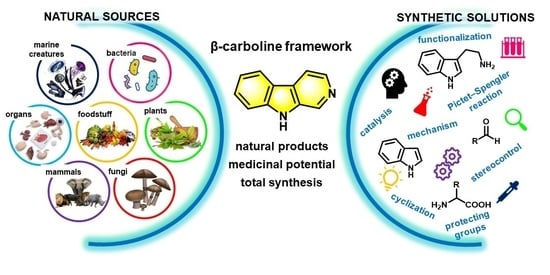
1. Introduction
Carbolines are a remarkable family of heterocyclic natural products, with outstanding pharmacological potential. They are determined by their tricyclic, pyridine-fused indole framework (where the rings are identified as A, B and C), and classified according to the degree of saturation (fully saturated: 1,2,3,4-tetrahydro; partially saturated: 3,4-dihydro; and unsaturated β-carbolines) and the position of the N-atom in the C-ring as α-, β-, γ- or δ-carbolines (Figure 1) [1,2]. β-Carboline alkaloids are widely distributed in nature including various plants, foodstuffs, marine creatures, insects, mammals as well as human tissues and body fluids. Numerous representatives of this family show various biological activities [3,4,5]. The fascinating diversity of structures and medicinal potential [6] inherent in them encourage several researchers to deal with the synthesis of β-carboline containing natural products and their synthetic derivatives [2,4,7,8,9]. Several commercial drugs or drug candidates such as vinpocetine, brovincamine, abecarnil, cipargamin, tadalafil, reserpine and lurbinectedin contain this unique framework (Figure 1).
This review focuses on the field of β-carboline research through selected examples from the last 5 years, where the key information on natural occurrence, structural diversity and biological activities is highlighted (Table 1). The comprehensive demonstration of recent advances in the synthesis of naturally occurring β-carbolines, including both simple β-carbolines (Section 2 of this review) and fused ring systems containing this framework (see Figure 1 and Section 3 of this review), is discussed with a special emphasis on the innovative framework construction steps.
2. Natural Products Containing a Simple β-Carboline Skeleton
A novel one-pot synthesis of norharman (1) and 1- or 6-substituted β-carbolines have been achieved by Wang and co-workers (Scheme 1) [109]. The designed synthesis starting from racemic tryptophan (74) and various amino acids (75) proceeds through sequential steps, driven by a reaction with I2 and TFA, followed by decarboxylation, deamination, Pictet–Spengler reaction and oxidation. By using various amino acids, further alkaloids and synthetic analogues were also obtained.
In 2015, Lood et al. reported the new enantiospecific gram-scale synthesis of (S)-eleagnine/(−)-tetrahydroharmane (2) (Scheme 2) [110]. Their synthesis started from the previously modified amino acid methyl ester 82, which included the appropriate chirality. The treatment of 82 with morpholine in the presence of t-BuMgCl resulted in morpholine amide 83. Amide 83 went through a subsequent lateral lithiation, which was followed by the addition of Boc-protected o-toluidine (84) to furnish ketone 85 in gram scale. After that, ketone 85 was treated with KHMDS, then the potassium enolate formed was quenched with ethyl bromoacetate, which resulted in a diastereomeric mixture of indoline γ-lactones (86a and 86b). Treating this diastereomeric mixture with sulfuric acid in MeOH facilitated a sequential lactone ring opening and Boc-cleavage to form indole 87. Hydrogenolysis of Pf-protecting group and treatment of the free amine with Na2CO3 base led to lactam 88. As a final step, LiAlH4 reducing agent was applied on 88 to provide (S)-eleagnine (2) in a gram-scale synthesis with an enantiomeric excess (e.e.) >99%.
Reddy et al. have accomplished the stereoselective synthesis of natural compounds (−)-tetrahydroharman (2), (−)-komaroidine (3), N-tetrahydroharman (4), (+)-1-ethyl-9-methyltetrahydro-β-carboline (5) and (+)-N-acetylkomaroidine (6) using Ellman’s sulfinamide [(R)-tert-butanesulfinamide, 91] [111] as a chiral auxiliary generally used for the preparation of chiral amines (Scheme 3) [112]. The use of this readily available reagent as the source of asymmetric induction to establish the stereochemical configuration of these alkaloids makes this approach convenient and practical. N-Sulfinylimines 92 were prepared with the reaction of 90 and (R)-tert-butanesulfinamide (91) in the presence of Ti(OEt)4. The addition of alkyl-Grignard reagent to 92 led to the corresponding sulfinamides 93 with the expected stereochemical outcome and with high diastereoselectivity. In this reaction, the occupation of the less hindered equatorial position in the six-membered transitional state resulted in a favorable attack from the same face (Scheme 3). The synthesis was continued with a reductive or acidic removal of both the benzyl group and the sulfinyl auxiliary to afford products 2–5. A final classical acetylation on the piperidine nitrogen atom was needed to prepare target molecule 6.
The chemical study on the marine tunicate Cnemidocarpa stolonifera contributed to the discovery and synthesis of 3 new alkaloids, including a β-carboline alkaloid called stolonine C (7, Scheme 4) [20]. The synthesis starts from l-tryptophan methyl ester (95) viaPictet–Spengler reaction with formaldehyde followed by dehydrogenation of the C-ring and hydrolysis of the ester group. Intermediate 96 was transformed into an amide with taurine resulting in final product 7.
Eudistomins are important natural products belonging to the family of β-carboline alkaloids, and show a wide variety of biological activities [26,113,114,115]. Kamal et al. have developed an efficient method for the synthesis of diverse β-carboline derivatives and naturally occurring alkaloids such as eudistomins I, N, T and U (8, 9, 10 and 11, respectively) [116] which are isolated from ascidians (Scheme 5) [23,24,27,116,117]. The protocol is based on the Pictet‒Spengler reaction of l-tryptophan (97) and the corresponding aldehydes, subsequent decarboxylative aromatization of the C-ring employing NCS and variation of classical synthesis methods in the final steps, providing an easier and more convenient pathway compared to the previously reported methods. In addition, the most efficient synthesis of 10 has been described using a one-pot approach.
It is worth mentioning that the NBS-promoted oxidative dehydrogenation of the C-ring has also been devised and has become very popular recently [118]. The method also provides access to a variety of β-carbolines and can be applied effectively in generating molecule libraries.
The beneficial pharmacological efficacy of eudistomins has prompted other researchers to further improve their total synthesis [115]. In 2015, also Kamal et al. reported a three-step synthesis of eudistomin U (10) starting from L-tryptophan methyl ester (95 ∙ HCl), which underwent a Pictet–Spengler condensation with N-acetylindole-3-carboxaldehyde (103) to form THβC ester 104 (Scheme 6). Ester hydrolysis and loss of the N-acetyl group of intermediate 104 provided carboxylic acid 105. Compound 105 went through an iodobenzene diacetate-mediated oxidative decarboxylation to afford 10 in 51% overall yield (Scheme 5) [119]. Using this method, the authors also developed the synthesis of eudistomin I (8) [119], where iodobenzene diacetate mediates not only decarboxylation and aromatization of the THβC ring, but also the partial dehydrogenation of the pyrrolidine ring in a one-pot operation.
An alternative synthesis of 10 through the reaction of tryptamine (108) and indole-3-carbonitrile (109) followed by dehydrogenation of intermediate 110 with palladium on carbon was described in two steps with 68% overall yield (Scheme 7) [120].
In 2016, Ábrányi-Balogh and co-workers developed a microwave-assisted one-pot protocol in the presence of T3P® for the preparation of substituted 3,4-dihydro-β-carbolines (DHβCs). The new method was also applied for the synthesis of isoeudistomin U alkaloid (12) starting from tryptamine (108) and indole-3-carboxylic acid (111) with 50% yield (Scheme 8) [121].
Gaikwad et al. have developed an iodine-mediated metal-free protocol for the chemoselective dehydrogenation and aromatization of THβCs into DHβCs and β-carbolines in the presence of H2O2 as the oxidant in DMSO (Scheme 9) [122]. The method was applicable not only for the preparation of variously substituted β-carbolines but also for that of bioactive natural products eudistomin U (10), harmalan (13), isoeudistomin M (14), norharman (1), harman (15) and kumujian C (16). The plausible reaction mechanism of the dearomatization protocol was also presented, which starts with a N-iodination followed by an N-dehydroiodination of THβCs 112 and 113, respectively. These two steps result in the imine (13 or 14), which is then further transformed into an ammonium intermediate (114) by an additional N-iodination. After deprotonation and dehydro-iodination, the corresponding β-carboline (1, 15, 116 or 117) is formed. The generated HI reacts with DMSO to give molecular iodine and dimethyl sulfide (DMS). The role of H2O2 is to speed up the reaction by the oxidation of DMS back into DMSO. Intermediates 116 and 117 could then be transformed into eudistiomin U (10) or kumujian C (16) in the final step.
Szabó et al. have recently published the first total synthesis of alkaloids trigonostemine A, B and G (17–19) and the preparation of pityriacitrin (20) and hyrtiosulawesine (21) (Scheme 10) [123,124]. The appropriate key intermediates kumujian C (16) and its derivatives (132, 133) were prepared via a novel and practical synthesis route. In the first step, the Pictet–Spengler reaction of tryptamines (108, 118, 119) and glyoxylic acid monohydrate gave THβC carboxylic acids (120–122). This was followed by the esterification of compounds 120–122. Aromatization of the C-ring was carried out in the presence of elemental sulfur. Afterwards the ester group of 126–128 was reduced to give alcohols. Finally, oxidation of the hydroxy group in alcohols 129–131 was achieved using MnO2 on Celite®, and aldehydes 16, 132, 133 were isolated in good yields. The synthesis of alkaloids 17–21 was then performed from these building blocks (16, 132, 133), which were reacted with indoles (134–137) to afford secondary alcohols and then oxidized with active MnO2 to provide the first total synthesis of trigonostemines A (17) and B (18), carried out in 7 steps. At the same time, this pathway represents a new synthetic method for the preparation of pityriacitrin (20). An additional step was needed to produce hyrtiosulawesine (21), where the target compound was obtained by O-demethylation of derivative 143 in pyridine hydrochloride. The first total synthesis of trigonostemine G (19) was performed in 8 steps analogously to the above method. The sensitive OH moiety of the indole was protected by a TBDMS group, which was removed under mild conditions in the final step with CsF (Scheme 10).
The IBX-mediated total synthesis of alangiobussine (22) and alangiobussinine (23) was achieved through the oxidative addition of isocyanide 146 to tryptoline (145) [125]. The general method (Scheme 11) represents an efficient preparation for imino-carboxamides, where tetrahydroisoquinolines and tryptolines were reacted with isocyanides. The IBX-mediated oxidative addition protocol was successfully applied for the gram-scale synthesis of natural product alangiobussine (22), which was further transformed to alangiobussinine (23) through the dehydrogenation of the C-ring in the presence of CuBr and DBU. The plausible reaction mechanism was also investigated by isotopic labeling and real-time NMR experiments, which made two pathways probable.
The first asymmetric total synthesis of haploscleridamine (24) was accomplished from histidine (Scheme 12) [126]. The synthesis starts with the tosylation of histidine methyl ester (152), then N-allylation with allyl bromide (154) was applied on intermediate 153 leading to compound 155, followed by a partial ester reduction to afford aldehyde 156. After Grignard reaction with vinylmagnesium bromide (159), alcohol 158 was formed, which was transformed into tetrahydropyridine congener 159 via ring-closing metathesis. After oxidation of alcohol to ketone 160, and reduction C=C double bond to intermediate 161, indole formation took place via Buchwald modification of the classical Fischer indole synthesis to obtain 163. Finally, 24 was synthesized by the reductive deprotection of 163 with Mg under ultrasonication conditions.
The first total synthesis of the potential anticancer agent, the β-carboline-vasicinone hybrid alkaloid (±)-peharmaline A (25) was accomplished in a short sequence starting from readily available starting materials (Scheme 13) [127]. Boc-protected pyrrolidone 164 was reacted with acyl chloride 165 in the presence of LiHMDS. After treatment with TFA, the compound thus formed (166) was reacted with 6-methoxytryptamine (119). The Pictet–Spengler reaction did not take place in a stereoselective manner: besides major diastereomer 167a, minor product 167b was also obtained. However, the inversion of the chiral center was observed in the minor diastereomer 167b during the next step. Natural product 25 was synthesized by N-acylation of the mixture of compounds 167a and 167b with reagent 168 and subsequent catalytic hydrogenation of nitro derivative 169.
Recently, Wang et al. [128] developed a new synthesis of komavine (26), a spiro-THβC derivative, and several analogous compounds via Pd-catalyzed cross-annulation of phenols (170) and tryptamine (108) with good functional group tolerance (Scheme 14). With this alternative protocol, THβC derivatives were synthesized with moderate to excellent yield through the C–O bond cleavage of phenols, C–H bond activation of tryptamines and C–N/C–C bond formation. The presented work offers an efficient protocol for converting phenolic lignin model monomers into valuable compounds.
A series of marine β-carboline alkaloids marinacarboline A–D (27–30) was achieved by Li et al. (Scheme 15) [57]. Their strategy applies classical synthetic methods starting from racemic tryptophan (74). First, esterification of 74 was implemented, then a Pictet‒Spengler reaction was performed to construct the β-carboline skeleton and the key intermediate (173) of the synthetic route. Amidation of 173 with the appropriate amines (174) resulted in marinacarbolines A (27) and C (29). For the synthesis of the other two marinacarbolines (28 and 30), the ester group of the key intermediate 173 was hydrolyzed, then transformed into an acid chloride (175), which was reacted with tyramine (176) or tryptamine (108) to achieve the synthesis of marinacarboline B (28) and D (30).
The simple total synthesis of water-soluble metatacarbolines A (31) and C–F (32–35) has been accomplished starting from β-carboline building blocks 177 [59]. Metatacarboline A was prepared in 3 steps by applying the Wittig reaction of aldehyde 177a with 178, followed by the catalytic reduction of the previously formed double bond and hydrolysis of ester groups to afford the desired product 31 (Scheme 16). Metatacarbolines C–F were produced in 5 steps starting from the protected aldehyde precursor 177b. First, the coupling of 177b with various amino acid esters was performed, then a formyl moiety was formed via acidic treatment of the acetal, followed by the same three steps described previously to form the other four metatacarbolines (32–35).
The first total synthesis of 6-hydroxymetatacarboline D (36) has been developed in 12 steps by Meng et al. via CuBr2-catalyzed mild oxidation method by the dehydrogenation of the DHβC’s C-ring (Scheme 17) [129]. The synthesis starts from 5-hydroxytryptophan (179), which was converted into Boc- and benzyl-protected compound 182 by applying protecting group chemistry. Compound 185 was obtained through deprotection and acylation. The β-carboline skeleton of 186 was formed via Bischler–Napieralski reaction, which was followed by the mild CuBr2-catalyzed conversion of DHβC 186 into β-carboline 187. The hydrolysis of ester 187 and the reaction with methyl l-prolinate followed by a catalytic debenzylation and amide formation with methyl l-threoninate led to the formation of 189. After all, double ester hydrolysis resulted in final product 36.
The total synthesis of shishijimicin A (37, Table 1), a marine natural product with remarkable antitumor activity has been described by Nicolau et al. in 48 steps (Scheme 18, Scheme 19, Scheme 20 and Scheme 21) [130]. Their method separates the natural product into three key intermediates. One of them, the β-carboline framework (191, Scheme 18) has been constructed in 8 steps by applying methods known from the literature [131] and functional group transformations to prepare the protected and in position 1 iodine-functionalized key intermediate (191).
Their convergent strategy for the construction of the complex structure of the target alkaloid 37 was continued with the synthesis of disaccharide aldehyde 198 where commercially available glucal (192) and glycosyl fluoride (196) were used as building blocks and transformed into 198 by using functional group chemistry and protecting group transformations (Scheme 19).
The synthesis of the required enediyne thioacetate precursor (212) started from key building block 199 proceeding in 19 steps with 25% overall yield (Scheme 20). The oxidation of oxime 199 with t-BuOCl resulted in a spontaneous [3+2] dipolar cycloaddition, which led to 200. This step was followed by a smooth deprotection and oxidation, then the key enediyne fragment 202 was added to the molecule. Removal of the MEM group followed by Swern oxidation and a further oxidation step furnished ketone 204. Horner−Wadsworth−Emmons olefination was performed, then intermediate 207 bearing a terminal acetylene bond was prepared through a sequence involving removal of the acetate group, cleavage of the TIPS moiety and silylation. Opening of the isoxazole ring was followed by the direct and stereoselective cyclization of 208 into 209 with the inversion of the configuration. The N-phthalide moiety was then converted to a methyl carbamate group in two steps to afford enediyne lactone 210. Reductive cleavage of the lactone function resulted in diol 211. Subsequent bis-silylation, selective cleavage of the primary OTMS moiety, Mitsunobu reaction, thioacetate formation and cleavage of the secondary OTMS group led to target molecule 212.
In the final steps, the β-carboline skeleton 131 was connected to the disaccharide 198, then various transformations were carried out to achieve intermediate 213 (Scheme 21). Finally, enediyne 212 and 213 were combined to complete the synthesis of shishijimicin A (37).
3. Fused Ring Systems Bearing a β-Carboline Skeleton
The reductive Pictet–Spengler reaction has been applied successfully for the preparation of canthine (38), the framework of biologically active canthine-6-ones, and harmicine (39) (Scheme 22) [120]. Instead of the commonly used aldehydes, nitriles (216, 217) were applied in the reductive intramolecular cyclization protocol, which was carried out with 10% Pd/C and H2. An additional dehydrogenation of the C-ring in the presence of 10% Pd/C catalyst was needed to form 38.
The synthetic route of (−)-harmicine ((−)-39) was performed as previously mentioned in part in Scheme 2, however with some differences (Scheme 23) [112]. N-Sulfinylimine was reacted with allylmagnesium bromide to afford the homoallylic sulfinamide with >99:1 ratio of diastereomers. In this reaction, a six-membered transition state also occurred, providing a high level of stereocontrol [132,133,134]. Compound 94c was prepared via base-catalyzed cyclization, then deprotection of the sulfinyl group and protection of the free amine with Boc2O furnished 219. Hydroboration of 219 resulted in alcohol 220, which was converted in the next step into its mesylate. Then deprotection of the Boc group and SN2 substitution of mesylate with a secondary amine took place. In the final step, debenzylation of compound 221 led to (−)-39.
The copper-catalyzed propargylation of cyclic aldimines was developed by Fandrick et al. (Scheme 24) [135]. By applying this procedure, (−)-(S)-harmicine ((−)-39) was synthesized from DHβC (147), using coupling agent 222, copper(II)isobutyrate and a chiral ligand to give intermediate 223, which was deprotected and transformed into (−)-39 in a titanium-catalyzed ring closure and subsequent C=N bond reduction with good yield and excellent stereoselectivity.
The one-step synthesis of racemic harmicine (39) has been recently reported by Nalikezhathu et al. (Scheme 25) [136]. The new method is based on the pyridyl-phosphine ruthenium(II)-catalyzed tandem Pictet‒Spengler reaction through the amination of alcohols. The β-carboline skeleton was effectively constructed providing several synthetic analogues. The subsequent cycloamination was also successfully applied for the synthesis of carbolines including harmicine 39, where its tetracyclic structure was built up in only one step from tryptamine (108) and 1,4-butanediol (224), in the presence of trifluoroacetic acid and Ru-catalyst (225).
The practical total synthesis of canthine derivative cordatanine (40) has also been accomplished (Scheme 26) [137] starting from tryptamine (108) and methoxymaleic anhydride (226), and by subsequent regioselective reduction of methoxymaleimide intermediate 227 into hydroxy derivative 228. Acid-catalyzed intramolecular dehydrative cyclization of 228 furnished compound 229. The methanolysis of lactam to ester and double oxidative aromatization of the C-ring took place in a one-pot reaction to give 230. The intramolecular cyclization in the presence of K2CO3 in MeOH led finally to cordatanine (40).
An alternate synthesis of cordatanine (40) has also been developed (Scheme 27) [138]. Tryptamine (108) was used again as the starting material which underwent a Pictet–Spengler reaction with ethyl glyoxalate (232), followed by direct oxidative aromatization catalyzed by Pd/C to give the synthetic intermediate kumujian A (232). Intermediate 232 was transformed into 233 via Claisen condensation. O-Methylation of 233 with methyl methanesulfonate in the presence of cesium carbonate resulted in a cis-trans mixture of 234. The final ring closure reaction was achieved by intramolecular amidation using NaH to obtain target molecule 40.
The first total synthesis of natural product griseofamine A (41) together with its diastereomer 16-epi-griseofamine A was described by Pan et al. (Scheme 28) [67]. The synthesis commences with the introduction of PMB group to commercially available 235 via reductive amination step to prepare 236. Then, a stereoselective Pictet–Spengler reaction was performed with the appropriate acetaldehyde for the construction of β-carboline skeleton. The reaction resulted in a mixture of diastereomers which were separated by column chromatography. The synthesis continued with the major diastereomer 237a. First, the Boc-protection took place to afford intermediate 238, which was further transformed with reagent 239 in a Suzuki–Miyaura cross-coupling reaction using Buchwald’s protocol with ligand 240. The linear prenylated product 241 went through multiple deprotection protocol (from 241 to 242). Finally, the tandem acylation/Lecay–Dieckmann condensation was performed avoiding the commonly used strong bases to achieve the first synthesis of 41. The diastereomer of 41, i.e., 16-epi-griseofamine A was prepared on the same reaction pathway with the slight difference that 237b was applied instead of 237a.
The construction of chiral β-carboline alkaloid (+)-deplancheine (42) has been achieved via (R)-SPINOL-TRIP-CPA-catalyzed asymmetric Pictet–Spengler reaction of indolyl dihydropyridine derivative 244 in two steps (Scheme 29) [54]. Intermediate 245 obtained with good enantiomeric excess was reduced with NaBH4 to obtain the desired product 42.
The enantioselective synthesis of (−)-geissoschizol (43) through Ir-catalyzed intramolecular ring closure as the key step has been accomplished by Zheng et al. (Scheme 30) [139]. The secondary allylic alcohol 247 was prepared from the reaction of protected tryptamine 246 and BuLi in the presence of acrolein. With key intermediate 247 in hand, the authors have attained a cyclization to forge the β-carboline skeleton. Intermediate 248 was prepared with Ir-catalyst in the presence of Zn(OTf)2 promoter and (S)-Carreira ligand in 62% yield with excellent e.e. The synthesis was continued with functional group transformations, aiming at the preparation of the corresponding aldehyde 250. Then, a Wittig reaction was performed to give 252, followed by deprotection and N-alkylation (from 253 to 255). The formation of D-ring has been implemented via Ni-catalyzed cyclization to form intermediate 255. Finally, deprotection of the indole nitrogen and reduction of the ester group of 257 resulted in target molecule 43.
The silent fungal Pictet–Spenglerase gene of Chaetomium globosum 1C51 residing in Epinephelus drummondhayi guts has been described to enable Pictet–Spengler reaction between 1-methyltryptophane and flavipin to give unnatural products containing novel skeletons in a natural way. Chaetogline alkaloids are representative compounds of this gene-implied strategy as a biosynthetic machinery to produce bioactive compounds [71]. Lei et al. have recently described the first total synthesis of alkaloids chaetogline C–F, among which chaetogline E (44) and F (45) are β-carbolines, derived from the genetically modified fungus via a divergent synthesis way (Scheme 31) [140]. The synthesis began with the commercially available 3,4,5-trimethoxybenzoic acid (258) to produce previously described lactone 259. After protecting group transformations, 260 was formed, which was transformed into 261 through aminolysis, oxidation and hydrolysis to achieve spontaneous cyclization. Condensation of 261 with 262 resulted the intermediate 263, which went through a Pictet–Spengler reaction and hydrolysis to give key intermediate 264 with the desired β-carboline skeleton. Further transformation of 264 led to chaetogline E (44) via global deprotection with AlCl3. For the synthesis of caethogline F (45), Barton decarboxylation was used to eliminate the carboxyl group and to provide 265. After that, 265 was oxidized, hydrolyzed and methylated to afford 266, which was deprotected with BBr3 and formed the desired product 45.
Natural products containing a 12H-pyrido-[1,2-a:3,4-b’]diindole framework like 6-oxofascaplysin (46) and fascaplysin (47) show a broad range of bioactivities. In 2018, Zhidkov et al. reported the new synthesis of 47 and the first preparation of 46 (Scheme 32) [141]. Their protocol starts with the reaction of unsubstituted indigo 267 and diethyl malonate (268) in the presence of NaH to give pentacycle 269. The subsequent hydrolysis and decarboxylation were carried out under reflux conditions in an excess of 40% hydrobromic acid to furnish 46 with 70% overall yield. Reduction of the keto moiety with BH3·THF complex, followed by hydrolysis and oxidation with air gave 47 in 43% isolated yield.
The one-pot synthesis of natural product evodiamine (48) and a variety of its closely related derivatives was achieved using a three-component biscyclization reaction (Scheme 33) [142]. The synthesis applied readily available substrates such as tryptamine (108) and N-methylisatoic anhydride (270) in the presence of TFAA and DABCO. With this method, a variety of polycyclic β-carboline scaffold-containing substrates has been synthesized, however as a limitation, a few products were only obtained in poor yield.
The novel synthesis of suaveoline alkaloids (49–51) was described by Zhao et al. applying a new method for pyridine incorporation starting from propargyl amines and unsaturated carbonyl compounds through a tandem reaction including condensation–alkyne isomerization–6π 3-azatriene electrocyclization sequence at the late stage of the synthesis (Scheme 34) [143]. The synthesis started from the readily available L-tryptophan methyl ester (95), which was converted into 271 according to previously described methods [144,145]. Next, aldehyde 271 was transformed with propargyl amine hydrochloride (272) into the desired pentacycle 273, including formation of a pyridine ring. This was followed by the cleavage of the benzyl group leading to norsuaveoline (49). An additional methylation step was performed on indole nitrogen of 273 before N-debenzylation to form the other target alkaloid suaveoline (50). To demonstrate the flexibility of the late-stage pyridine incorporation, the authors made a reaction with 271 using another propargyl amine counterpart (274). The desired product 275 was further converted into macrophylline (51) through a Pd(OH)2-catalyzed debenzylation and methylation of the piperidine nitrogen atom.
Smith et al. elaborated a novel asymmetric propargylation as a new approach for the preparation of β-carboline alkaloids, highlighted by the formal synthesis of (–)-decarbomethoxy-dihydrogambirtannine (52) and (+)-strictamine (53) (Scheme 35) [146]. Starting from DHβC (147), the propargylation was accomplished with allenylboronic acid pinacol ester (276) under asymmetric conditions to form 277. To obtain 278, 277 was reacted with TFA and 6-bromomethyl-2-pyrone. Finally, an MW-assisted cyclization was performed to complete the synthesis of target molecule 52. For the synthesis of 53, 277 went through further transformations and an Au(I)/Ag(I)-based 6-endo-dig cyclization to afford the core framework in compound 281. According to previously described transformations by Zhu and co-workers [147], alkylation and Ni-promoted cyclization in the presence of Et3SiH led to 53 with poor yield.
Formal total synthesis of (±)-strictamine (53) based on gold-catalyzed cyclization was developed by a Japanese research group (Scheme 36) [148]. Starting from readily available tryptamine (108), a formylation with HCO2Et and cyclization with POCl3 was performed to give DHβC (147), followed by Grignard reactions and protecting group chemistry to afford 287 [149]. The key intermediate tetracyclic indolenine 289 was formed via SPhosAuCl (288)-promoted cyclization to construct the A–D ring system of akuammiline alkaloids. Functional group modifications on 289 operating with a Boc protecting group and ester group formation followed by an N-alkylation led to key intermediate 282. After that, it is possible to prepare 53, based on a previous publication [147].
The synthesis of monoterpene indole alkaloid (±)-arbornamine (54) was described in only 6 steps (Scheme 37) [150]. First, benzyl-protected tryptamine 294 was reacted with dimethyl ester 295, resulting in intermediate 298. Debenzylation was carried out under catalytic hydrogenation conditions to afford 297, which was further transformed into 298 with (Z)-1-bromo-2-iodobut-2-ene (254). A general selenylation-elimination protocol resulted in the δ-lactam containing product 299. The closure of the final ring was achieved via reductive Heck cyclization promoted by Ni(cod)2, leading to the appropriate pentacyclic product 300. In the final stage, reduction of the ester moiety of 300 provided the desired alkaloid 54.
(+)-Tacamonine (55), containing a pentacyclic framework, as well as its closely related bioactive derivatives like eburnamine [151] or vincamine [152] are considered as pharmacologically promising molecules. The first synthesis of (±)-tacamonine containing 3 stereocenters has been achieved by Massiot and co-workers [153]. Recently, the asymmetric synthesis of (+)-tacamonine (55) has been reported involving a stereoselective radical cyclization as the key step (Scheme 38) [154]. The synthesis starts from a commercially available oxazolidinone auxiliary (301) through an acylation followed by an allylation to form intermediate 302. After that, 302 was modified with the removal of oxazolidinone part followed by a cross-metathesis with ethyl acrylate (303) resulting in chiral acid 304. Next, intermediate 304 was reacted with DHβC (147) using DCC and HOBt, followed by the reaction with PhSH in the presence of BF3·OEt2 to afford α-phenylsulfanyl amide. The precursor for radical cyclization was provided by Boc-protection of the indole nitrogen atom. Then, 305 was treated with Bu3SnH and ACCN to form 306 which went through the thermolytic removal of the Boc group, then the reduction of the ester followed by an oxidation step with concomitant cyclization to provide 55.
In 2018, alkaloids containing the complete geissoschizine skeleton were synthesized via bioinspired oxidative cyclization, which led to the selective formation of the N4-C16 bond [155]. In this research, (−)-17-nor-excelsinidine (56) was synthesized (Scheme 39), including an α-electrophilic chlorination of a 7-membered-ring lactam and its ring rearrangement via nucleophilic substitution. Two oxidative cyclization strategies were developed to construct (−)-17-nor-excelsinidine starting from enantiopure d-trypthophan (D–73), which was transformed into 307 via Pictet–Spengler reaction followed by a protecting maneuver. After this, Mannich addition with 308 and N-alkylation with 1-bromo-2-iodobut-2-ene (254) were accomplished to give 309. In the presence of Ni(cod)2, radical cyclization of 309 occurred, then removal of the benzyl ester resulted in key intermediate 311 containing the required scaffold. Next, the oxidative cyclization took place which was achieved on two alternative pathways (Scheme 39A,B) to produce alkaloid 56. Following the reaction pathway A, the indole nitrogen and the carbonyl of the ester of 311 were connected by forming 7-membered-lactam 312. The oxidative process was accomplished in two stages. First, the C16-carbon was subjected to umpolung by diastereoselective chlorination to obtain 313. This was followed by the addition of sodium carbonate in aqueous methanol was induced the nucleophilic substitution of the chlorine by the N4-nitrogen and the cleavage of the lactam to form enantiopure (−)-56. Choosing another synthetic route B, 311 was transformed into (+)-geissoschizine (57) through a formylation step. After that, 57 was deprotonated with KHMDS and the resulted dianion was then submitted to oxidation with iodine leading to (−)-56 methyl ester, which was then saponified to 56.
The same authors reported the total synthesis of further 3 closely related mavacuran alkaloids: (+)-16-epi-pleiocarpamine (58), (+)-taberdivarine H (59) and (+)-16-hydroxymethyl-pleiocarpamine (60) by applying their method described above for the construction of geissoschizine skeleton (Scheme 40 and Scheme 41) [156]. The synthesis of 58 starts with the formylation step of 311 leading to the formation of 57. Heating of 57 with PMB-Br in MeCN resulted in quaternary ammonium compound 314. In the following reaction step, deprotonation of the indole NH of 314 followed by the addition of I2 gave a mixture of 315 and 316 containing the desired mavacuran skeleton. Finally, dequaternization of 316 with TMSCl furnished the target molecule 58, although with a poor yield (Scheme 40).
Similarly to the method described above, the basic framework of the target molecules was built by applying the key oxidative coupling induced by KHMDS in the presence of I2 as this latter masks the nucleophilicity of the aliphatic nitrogen center and locks it in the suitable cis configuration leading to intermediates 318 and 319 (Scheme 41). (+)-Taberdivarine H (59) was synthesized by quaternization of the tertiary nitrogen atom of 317 with MeI followed by an oxidative coupling to give 318, and subsequent ester hydrolysis and selective decarboxylation. The synthesis continued with the oxidative coupling of 319 and deprotection of 320 with BBr3. Selective Krapcho decarboxylation of diester 321 occurred to form compound 58. Selective reduction of key intermediate 321 with DIBAL resulted in the desymmetrized product 322, followed by a deformylation step in the presence of NaBH4 to accomplish the synthesis of 60. Compound 60 was transformed into 58 with NaH in boiling toluene and by subsequent addition of water.
The total synthesis of C-mavacurine alkaloids (±)-pleiocarpamine (61), (±)-normavacurine (62) and (±)-C-mavacurine (63) was accomplished by Sato et al. (Scheme 42) [157]. The innovative key step of the synthesis is a metal carbenoid cyclization by an N–H insertion within a Corynanthe-type compound bearing a diazo function, where the molecular conformation is fixed by an amine-borane complex. The synthesis starts with the preparation of α,β-unsaturated ester 323 from tryptamine (108), applying the previously reported protocol by Vincent and co-workers [155]. This was followed by the Ni(cod)2-mediated reductive cyclization to afford a mixture of trans-324 and the desired cis-324. After that, trans-324 was treated with t-BuOCl, then with HCl in MeOH to form an iminium intermediate, which was effectively transformed into cis-324 in the presence of Noyori-type catalyst, formic acid and Et3N in DMF solvent. Next, cis-324 was treated with LDA and methyl formate to obtain the D-ring containing (±)-geissoschizine (57). Natural product 57 was converted into the key intermediate of the reaction, diazo compound 325 using tosyl azide in the presence of Et3N. As an essential compound for the further steps, 325 was transformed into borane-complex 326 with BH3·THF to fix the appropriate conformation for the next steps. The cyclization precursor 326 underwent a carbene-generated cyclization with catalyst 327 to form the E-ring containing derivative 328. The amine-borane bond was easily cleaved by heating in the presence of trimethylamine oxide to obtain (±)-pleiocarpamine (61) and intermediate 329. For the synthesis of (±)-normavacurine (62), 329 was used in a reduction step carried out with LiAlH4 andfurther transformation into (±)-C-mavacurine (63) by treatment with an excess of MeI.
The C19-oxo-functionalized eburnane alkaloids (64–67) were synthesized by Trost et al. (Scheme 43) [158]. The synthesis involves a Pd-catalyzed asymmetric allylic alkylation step of an N-alkyl-α,β-unsaturated lactam as a key step for the further ring system construction. Using Corey and Baran’s method [159], Boc-protected tryptamine (330) and aldehyde 331 were transformed into tryptamine derivative 332. After that, amine 332 was reacted with tert-butyl malonate (333) in the presence of Mukaiyama’s reagent (334) to perform an amidation reaction. The formed intermediate 335 was treated with FeCl3 on silica via ketal cleavage and cyclization to provide the α,β-unsaturated lactam 336. The reaction of intermediate 336 with nucleophile 337, i.e., the key step of the synthesis was accomplished applying CpPd(cinnamoyl) as a catalyst, Trost ligand (338) and Barton’s base as an additive, giving 75% yield with e.e., 90%. The key intermediate 339 thus obtained underwent a desilylation in the presence of TBAF, followed by a simultaneous Boc-group removal and transesterification to afford 340. In the next step, the formation of the C-ring was accomplished through a Bischler–Napieralski reaction using Movassaghi’s protocol to prepare 341 [160]. The exposure of 341 to excess of MeLi led to the corresponding ketone 342, where no by-product was observed because of the intramolecular chelate formation. This was followed by the chemoselective dihydroxylation of terminal olefin bond in the presence of OsO4 and NMO, then by the reaction with NaIO4 on silica leading to an aldehyde intermediate, which directly cyclized to give pentacycle 343. From this point, the synthesis was continued via divergent routes to the target molecules. First, hydrogenation took place, where the saturation of the double bond of the tetrahydropyridine ring and concomitant partial reduction of the C19 carbonyl group resulted in a mixture of (+)-19-oxoeburnamine (64) and 19-hydroxyeburnamine (65). Focusing on the other two target molecules, a Ley–Griffith oxidation was performed on 343, which led to compound 344. The selective reduction of C19-ketone 344 was performed in the presence of l-selectride to obtain 19-(S)-hydroxy-Δ14-vincamone (66). Finally, hydrogenation of 66 using 10% PtO2 resulted in (−)-19-hydroxy-Δ14-eburnamonine (67).
The total synthesis of (+)-vallesamidine (68) in 23 steps and that of (+)-14,15-dehydrostrempeliopine (69) in 24 steps were completed by Anderson and co-workers through an asymmetric synthesis using the tetracyclic key intermediate 359 (Scheme 44) [161]. For the preparation of target molecules, the synthesis starts with asymmetric Michael addition with 2-bromonitrostyrene 345 using chiral Ni(II)-complex 346 as the catalyst [162]. Next, a nitro-Mannich and lactamization cascade was accomplished, which continued with a Krapcho decarboxylation to give the nitro lactam 348. Then, a palladium-catalyzed Tsuji–Trost allylation was performed, which was followed by a nitro reduction and an intramolecular C–N coupling to construct the basic tricyclic β-carboline skeleton on gram scale (from 348 to 350). After that, 350 underwent an in situ thionium ion formation by the sequential addition of Lawesson’s reagent, MeI and NaBH4. Protonation of the basic piperidine nitrogen with trifluoroacetic acid and ozonolysis gave the aldehyde, which was treated with allyl chloroformate to obtain 352. Subsequent treatment of allyl carbamate 352 with catalytic Pd(PPh3)4 induced the decarboxylative allylation to give 353. This was followed by the Knoevenagel condensation of 353 with dimethyl malonate to afford the α,β-unsaturated malonate 354. Using precursor 354 in the reaction with Yb(OTf)3, a Lewis acid-catalyzed [1,4]-hydride transfer and Mannich-type cascade cyclization was performed, which resulted in compound 355 containing the appropriate E-ring. The D-ring with the C14=C15 double bond was installed via alkene formation through diastereoselective ring-closing metathesis (from 355 to 358). To form the metathesis precursor from 355, the reduction of this malonate to the corresponding diol was carried out. The diol was selectively protected, then the oxidation of the other OH-group was performed, which was followed by an olefination and the removal of the protecting group. After that, Swern oxidation and Wittig reaction took place to furnish the appropriate trialkene. 358. Then 358 was introduced to the Hoveyda–Grubbs catalyst and the subsequent metathesis reaction occurred to afford key intermediate 359. From intermediate 359, the target molecules were formed via two different pathways: 68 was obtained after simple functional group transformations, while 69 was formed by further functionalization and ring closure reaction of 360.
The enantioselective total synthesis of (+)-peganumine A (70) was achieved by Zhu and co-workers in 7 steps by applying multiple bond formation methods (Scheme 45) [163]. The synthesis of the octacyclic complex natural product starts from readily available 6-methoxytryptamine (118). By combining functional group transformation and a Liebeskind–Srogl cross-coupling (361 into 364), this starting material was transformed into advanced intermediate 365. Further reactions from 365 led to the construction of tetracyclic intermediate 367 via one-pot C–C bond forming lactamization and transannular condensation. Finally, a one-pot chiral (S)-368 thiourea/benzoic acid-catalyzed domino reaction merged the two achiral building blocks (118, 367) into the octacyclic compound (70) via enantioselective Pictet–Spengler reaction followed by TFA-catalyzed transannular cyclization.
Reserpine (71) is a well-known drug for the treatment of high blood pressure. From the first total synthesis of 71 reported by Woodward and co-workers, [164] numerous synthetic approaches have been reported [165]. Recently, a new total synthesis of reserpine (71) has been accomplished using a desymmetrization protocol, which applied a simplified nonstereogenic E-ring precursor [166]. The developed strategy constitutes an unconventional form of remote stereoinduction. Late-stage functionalization of the reserpine E-ring, including a contra-steric C17 oxygenation and a stereoselective C18 ketone reduction, also played pivotal roles in completing the synthesis. As shown in Scheme 46, the synthesis began with the preparation of the bicyclic ketoaldehyde (372) through a reductive [4+3] cycloaddition between acetoxyfulvene (369) and 1,1,3,3-tetrabromoacetone (370) affording bicyclic enolacetate 371, followed by an enolacetate hydrolysis with aldehyde epimerization. One-carbon homologation of 372 was performed by engaging its aldehyde functionality resulting in the methyl enol ether mixture of geometrical isomers (373) as an E-ring precursor. This was followed by an improved Pictet–Spengler protocol in the reaction with tryptamine derivative 118 to construct the THβC skeleton-containing compound 374. The N atoms of 374 were protected with Cbz and Boc. At this stage, a desymmetrization protocol was performed starting with an OsO4-catalyzed alkene dihydroxylation in the presence of K3Fe(CN)6 to afford diol 375. Intermediate 375 was then treated with Pb(OAc)4 to form a mixture where the bis-hemiacetal 376 was the predominant compound instead of its parent dialdehyde. This mixture went through a Pd/C-mediated hydrogenation, and Cbz group removal where the only detectable product was dialdehyde 377. Next, an intramolecular iminium formation took place induced by an in-situ hydrogenation resulted 378. After that, the irreversible dehydrogenation and the single stereoisomer suggested the iminium formation took place by selectively engaging one of the two diastereotopic aldehyde sin-379. Intermediate 379 went through various transformations including Pinnick oxidation and esterification, HOAc catalyzed epimerization and re-protecting with Boc2O, resulting in 380, which was followed by a regio- and stereoselective C17 oxygenation with L-proline/nitrosobenzene to complete the substitution pattern required for the E-ring. Pummerer-type conditions (DMSO, Ac2O) were applied to furnish methylthio methyl ether intermediate 381, which underwent a reductive desulfurization to prepare the oxygen methylated alcohol 382. To complete the synthesis, 382 was reacted with 3,4,5-trimethoxybenzoyl chloride (383) and removal of the Boc-carbamate by heating in the presence of silica gel to achieve the target molecule reserpine (71).
The first enantioselective synthesis of glycosylated monoterpenoid alkaloids (–)-5-carboxystrictosidine (72, Scheme 47) and (−)-rubenine (73, Scheme 48) was accomplished by Rakamitsu et. al. [167]. The innovative part of this publication is a sequential anti-selective organocatalytic Michael reaction continued with a Fukuyama reduction and finished by a cyclization to form an optically active dihydropyran ring. The first stage of the synthesis targeted the preparation of hemiacetal 390. First, thioester derivative 386 was prepared by Knoevenagel condensation from the readily available 3-trimethylsilylpropanal (384) and reagent 385, applying a known literature method [168]. This was followed by the innovative key step, the anti-selective Michael reaction, which was finally performed with organocatalyst 387 and thioaldehyde 388 in Et2O solvent. The formation of the appropriate dihydropyran ring-containing key intermediate 390 was finally achieved applying Fukuyama’s protocol with Pd/C and Et3SiH, followed by a spontaneous cyclization. Next the glycosylation of hemiacetal diastereomeric mixture 390 was examined through the reaction with imidate 391. The key issue of this step was to prepare 392 with controlled stereoselectivity. The silyl group removal was accomplished with TBAT, thereafter the hydroboration/oxidation was performed with Schwartz’s reagent (Cp2ZrClH) in the presence of 9-BNN. In this reaction the thioether was also oxidized to sulfoxide 393, which was essential for the next sulfoxide elimination reaction. To prepare the terminal olefin bond containing moiety in 394, Tietze’s sulfoxide elimination protocol [169] was used which resulted in another key molecule of the synthesis route called secologanin tetraacetate (394). To complete the total synthesis of 72, a diastereoselective Pictet–Spengler reaction was performed [170] with 95. Finally, hydrolysis of the acetate groups of 395 led to target molecule 72.
In the same publication, the authors used key intermediate 394 also for the first total synthesis of (−)-rubenine (73, Scheme 48). Initially, aldehyde 394 was converted into cyclic acetal 397 upon reaction with 1,2-phenylenedimethanol (396). Then, the diastereoselective epoxidation and acetal removal of 397 was accomplished in the reaction with m-CPBA in HFIP solvent to obtain intermediate 398. Next, the Pictet–Spengler reaction of 395 with 95 generated the desired 3S-isomer 399. The E-ring formation was induced by the direct loading of the diastereomeric mixture on silica gel to obtain intermediate 400. The synthesis of (−)-rubenine (73) was completed by the removal of the acetate moieties by methanolysis, followed by the highly strained d-ring formation upon basic lactonization reaction.
4. Conclusions
This review summarizes the recently reported syntheses of naturally occurring β-carbolines and closely related synthetic derivatives through the selected examples of 73 natural products. It is evident that over the last 5 years this field of alkaloid chemistry has experienced many new and novel synthetic routes, including innovative strategies for the construction of the title framework. Not only the applied methods and synthesized products are interesting for organic chemists, but they also represent a remarkable pharmacological potential. The urgent need for new drug candidates is the driving force of the development highlighted in this review. We believe that the presented synthetic methods give a valuable insight into β-carboline chemistry, while providing inspiration and encouraging further explorations in this family of compounds.
Author Contributions
Conceptualization, T.S., B.V., M.M.; literature search, T.S.; writing—original draft preparation, T.S.; writing—review and editing, B.V., M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
List of Abbreviations and Named reagents.
| 9-BNN | 9-borabicyclo[3.3.1]nonane |
| Å | angstrom |
| Ac | acetyl |
| ACCN | 1,1′-azobis(cyclohexanecarbo-nitrile) |
| AIBN | azobisisobutyronitrile |
| Alloc | allyloxycarbonyl |
| aq | aqueous |
| Barton’s base | 2-tert-butyl-1,1,3,3-tetra-methylguanidine |
| Bn | benzyl |
| Boc | tert-butyloxycarbonyl |
| brsm | yield based on recovered starting material |
| BuLi | n-butyllithium |
| Bz | benzoyl |
| CAN | cerium ammonium nitrate |
| cap | caprolactamate |
| CDI | carbonyldiimidazole |
| c-Hex2BH | dicyclohexylborane |
| cod | cyclooctadiene |
| conc. | concentrated |
| Cp2TiMe2 | bis(η5-cyclopentadienyl)-dimethyl-titanium |
| CuDPP | copper(I)-diphenyl phosphinate |
| d | day |
| d.r. | diastereomeric ratio |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| dba | dibenzylideneacetone |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DBN | 1,5-diazabicyclo[4.3.0]non-5-ene |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DEAD | diethyl azodicarboxylate |
| DHβC | 3,4-dihydro-β-carboline |
| DIBAL | diisobutylaluminum hydride |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-dimethylaminopyridine |
| DME | 1,2-dimethoxyethane |
| DMF | dimethylformamide |
| DMP | Dess–Martin periodinane |
| DMS | dimethyl sulfide |
| DMSO | dimethyl sulfoxide |
| e.e. | enantiomeric excess |
| e.r. | enantiomeric ratio |
| EDCl | 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide |
| Et | ethyl |
| h | hour |
| HFIP | 1,1,1,3,3,3-hexafluoroisopropyl alcohol |
| HOBt | 1-hydroxybenzotriazole |
| IBA | 2-iodosobenzoic acid |
| IBX | 2-iodoxybenzoic acid |
| KHMDS | potassium bis(trimethylsilyl)-amide |
| Lawesson’s reagent | 2,4-bis(4-methoxy-phenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide |
| LDA | lithium diisopropylamide |
| LiHMDS | lithium bis(trimethylsilyl)amide |
| L-selectride | lithium tri-sec-butyl(hydrido)-borate |
| m-CPBA | meta-chloroperbenzoic acid |
| Me | methyl |
| MEM | 2-methoxyethoxymethyl ether |
| min. | minute |
| MS | molecular sieve |
| MW | microwave |
| Nap | 2-naphthylmethyl ether |
| NB | 2-nitrobenzyl |
| NBS | N-bromosuccinimide |
| NCS | N-chlorosuccinimide |
| NMM | N-methylmorpholine |
| NMO | 4-methylmorpholine N-oxide |
| NMP | N-methyl-2-pyrrolidone |
| Ns | 2-nitrobenzenesulfonyl |
| o-NBOH | o-nitrobenzylalcohol |
| Oxone® | 2 KHSO5·KHSO4·K2SO4 |
| PDC | pyridinium dichromate |
| Pf | phenylfluorenyl |
| Phth | phthaloyl |
| pin | pinacolato |
| PMB | p-methoxybenzyl |
| Pr | propyl |
| psi | pound-force per square inch |
| Py | pyridine |
| r.t. | room temperature |
| Schwartz’s reagent | chloridobis(η5-cyclo-pentadienyl)hydridozirconium |
| T3P® | propylphosphonic anhydride |
| TBAF | tetrabutylammonium fluoride |
| TBAT | tetrabutylammonium difluoro-triphenylsilicate |
| TBDPS | tert-butyldiphenylsilyl |
| TBDMS | tert-butyldimethylsilyl |
| t-Bu | tert-butyl |
| TES | triethylsilane |
| TMG | tetramethylguanidine |
| Tf | trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| TFAA | trifluoroacetic anhydride |
| THF | tetrahydrofuran |
| THβC | 1,2,3,4-tetrahydro-β-carboline |
| TIPS | triisopropylsilyl |
| TMG | 1,1,3,3-tetramethylguanidine |
| TMP | 2,2,6,6-tetramethylpiperidine |
| TMS | trimethylsilyl |
| TPAP | tetrapropylammonium perruthenate |
| Ts | p-toluenesulfonyl (tosyl) |
| Tsdpen | N-tosyl-1,2-diphenylethylene-1,2-diamine |
References
- Smirnova, O.B.; Golovko, T.V.; Granik, V.G. Carbolines. Part 2: Comparison of some of the properties of α-, γ-, and δ-carbolines. Pharm. Chem. J. 2011, 45, 389–400. [Google Scholar] [CrossRef]
- Singh, V.; Batra, S. 1-Formyl-9H-β-carboline: A useful scaffold for synthesizing substituted and fused β-carbolines. Curr. Org. Synth. 2012, 9, 513–528. [Google Scholar] [CrossRef]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive β-carbolines in food: A review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Dan, W.; Schneider, U.; Wang, J. β-Carboline alkaloid monomers and dimers: Occurrence, structural diversity, and biological activities. Eur. J. Med. Chem. 2018, 157, 622–656. [Google Scholar] [CrossRef] [PubMed]
- Casal, S. Chapter 20. Potential effects of β-carbolines on human health. In Coffee: Consumption and Health Implications; The Royal Society of Chemistry: Croydon, UK, 2019; pp. 461–468. [Google Scholar]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. b-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef]
- Maity, P.; Adhikari, D.; Jana, A.K. An overview on synthetic entries to tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar] [CrossRef]
- Domínguez, G.; Pérez-Castells, J. Chemistry of β-carbolines as synthetic intermediates. Eur. J. Org. Chem. 2011, 2011, 7243–7253. [Google Scholar] [CrossRef]
- Devi, N.; Kumar, S.; Pandey, S.K.; Singh, V. 1(3)-Formyl-β-carbolines: Potential aldo-X precursors for the synthesis of β-carboline-based molecular architectures. Asian J. Org. Chem. 2018, 7, 6–36. [Google Scholar] [CrossRef]
- Inoue, S.; Okada, K.; Tanino, H.; Kakoi, H.; Goto, T. Trace characterization of the fluorescent substances of a dinoflagellate, Noctiluca miliaris. Chem. Lett. 1980, 9, 297–298. [Google Scholar] [CrossRef] [Green Version]
- Herraiz, T. Identification and occurrence of β-carboline alkaloids in raisins and inhibition of monoamine oxidase (MAO). J. Agric. Food. Chem. 2007, 55, 8534–8540. [Google Scholar] [CrossRef]
- Zhu, Y.-Y.; Li, X.; Yu, H.-Y.; Xiong, Y.-F.; Zhang, P.; Pi, H.-F.; Ruan, H.-L. Alkaloids from the bulbs of Lycoris longituba and their neuroprotective and acetylcholinesterase inhibitory activities. Arch. Pharmacal Res. 2015, 38, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-P.; Zou, Z.-X.; Zhang, Y.; Li, J.; Cheng, F.; Xu, P.-S.; Zhou, G.; Li, X.-M.; Xu, K.-P.; Tan, G.-S. New adenine analogues and a pyrrole alkaloid from Selaginella delicatula. Nat. Prod. Res. 2019, 33, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, N.; Bai, Z.; Di, J.; Zhang, H.; Dong, P.; Zhang, P. Chemical constituent from the peel of Trichosanthes kirilowii Maxim and their NF-κB inhibitory activity. Nat. Prod. Res. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.M.; Court, W.E. Leaf alkaloids of Rauwolfia caffra. Phytochemistry 1983, 22, 2297–2300. [Google Scholar] [CrossRef]
- Schumacher, R.W.; Davidson, B.S. Didemnolines A-D, new N9-substituted β-carbolines from the marine ascidian Didemnum sp. Tetrahedron 1995, 51, 10125–10130. [Google Scholar] [CrossRef]
- Rajemiarimoelisoa, C.F.; Boyère, C.; Pellissier, L.; Peuchmaur, M.; Randrianarivo, H.R.; Rakoto, D.A.D.; Jeannoda, V.L.; Boumendjel, A. Chemical composition of the pods of Albizia polyphylla. Nat. Prod. Res. 2016, 30, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Tulyaganov, T.; Kozimova, N.; Allaberdiev, F.K. Alkaloids from plants of the genus Nitraria. Chem. Nat. Compd. 2006, 42, 198–200. [Google Scholar] [CrossRef]
- Badger, G.M.; Beecham, A.F. Isolation of tetrahydroharman from Petalostyles labicheoides. Nature 1951, 168, 517–518. [Google Scholar] [CrossRef]
- Tran, T.D.; Pham, N.B.; Ekins, M.; Hooper, J.N.A.; Quinn, R.J. Isolation and total synthesis of stolonines A–C, unique taurine amides from the australian marine tunicate Cnemidocarpa stolonifera. Mar. Drugs 2015, 13, 4556–4575. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.B.; Saboune, H.; Abramowski, Z.; Towers, G.H.; Rinehart, K.L., Jr. The photoactive antimicrobial properties of eudistomins from the caribbean tunicate Eudistoma olivaceum. Photochem. Photobiol. 1988, 47, 377–381. [Google Scholar] [CrossRef]
- Tarzi, O.I.; Erra-Balsells, R. Photochemistry of the alkaloids eudistomin N (6-bromo-nor-harmane) and eudistomin O (8-bromo-nor-harmane) and other bromo-β-carbolines. J. Photochem. Photobiol. B Biol. 2005, 80, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Badre, A.; Boulanger, A.; Abou-Mansour, E.; Banaigs, B.; Combaut, G.; Francisco, C. Eudistomin U and isoeudistomin U, new alkaloids from the carribean ascidian Lissoclinum fragile. J. Nat. Prod. 1994, 57, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Kinzer, K.F.; Cardellina, J.H. Three new β-carbolines from the bermudian tunicate Eudistoma olivaceum. Tetrahedron Lett. 1987, 28, 925–926. [Google Scholar] [CrossRef]
- Rajesh, R.P.; Murugan, A. Spectroscopic identification of brominated, non-brominated alkaloids and evaluation of antimicrobial activity of Eudistomin-I, Eudistomin H, from green ascidian Eudistoma viride. J. Appl. Pharm. Sci. 2019, 9, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Rinehart, K.L., Jr. Eudistomins A, D, G, H, I, J, M, N, O, P, and Q, bromo, hydroxy, pyrrolyl and iminoazepino. beta-Carbolines from the antiviral caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1526–1528. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A-Q,. beta-Carbolines from the antiviral caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Kim, D.-C.; Quang, T.H.; Yoon, C.-S.; Ngan, N.T.T.; Lim, S.-I.; Lee, S.-Y.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory activities of indole alkaloids from kanjang (Korean fermented soy source) in lipopolysaccharide-induced BV2 microglial cells. Food Chem. 2016, 213, 69–75. [Google Scholar] [CrossRef]
- Picker, K.; Ritchie, E.; Taylor, W. The chemical constituents of australian Flindersia species. XXI. An examination of the bark and the leaves of F. laevicarpa. Aust. J. Chem. 1976, 29, 2023–2036. [Google Scholar] [CrossRef]
- Guo, E.; Hu, Y.; Du, T.; Zhu, H.; Chen, L.; Qu, W.; Zhang, J.; Xie, N.; Liu, W.; Feng, F.; et al. Effects of Picrasma quassioides and its active constituents on Alzheimer’s disease in vitro and in vivo. Bioorg. Chem. 2019, 92, 103258. [Google Scholar] [CrossRef]
- Shi, C.-C.; Chen, S.-Y.; Wang, G.-J.; Liao, J.-F.; Chen, C.-F. Vasorelaxant effect of harman. Eur. J. Pharmacol. 2000, 390, 319–325. [Google Scholar] [CrossRef]
- Miralles, A.; Esteban, S.; Sastre-Coll, A.; Moranta, D.; Asensio, V.J.; García-Sevilla, J.A. High-affinity binding of β-carbolines to imidazoline I2B receptors and MAO-A in rat tissues: Norharman blocks the effect of morphine withdrawal on DOPA/noradrenaline synthesis in the brain. Eur. J. Pharmacol. 2005, 518, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moty, S.; Sakai, S.; Aimi, N.; Takayama, H.; Kitajima, M.; El-Shorbagi, A.; Ahmed, A.; Omar, N. Synthesis of cytotoxic 1-polyhydroxyalkyl-β-carboline derivatives. Eur. J. Med. Chem. 1998, 32, 1009–1017. [Google Scholar] [CrossRef]
- Sung, Y.; Koike, K.; Nikaido, T.; Ohmoto, T.; Sankawa, U. Inhibitors of cyclic AMP phosphodiesterase in Picrasma quassioides Bennet, and inhibitory activities of related beta-carboline alkaloids. Chem. Pharm. Bull. 1984, 32, 1872–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.H.; Gao, H.; Zhao, F.; Lin, H.W.; Pan, Y.M.; Zhou, G.X.; Yao, X.S. Anti-inflammatory alkaloids from the stems of Picrasma quassioides Bennet. Chem. Pharm. Bull. 2011, 59, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Li, H.; Luan, R.; Huang, G.; Liu, Y.; Wang, M.; Chao, Q.; Wang, L.; Li, D.; Fan, H.; et al. Identification of β-carboline and canthinone alkaloids as anti-inflammatory agents but with different inhibitory profile on the expression of iNOS and COX-2 in lipopolysaccharide-activated RAW 264.7 macrophages. J. Nat. Med. 2019, 73, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-F.; Zhang, Y.; Li, Y.; Li, X.-R.; Kong, L.-M.; Tan, C.-J.; Li, S.-L.; Di, Y.-T.; He, H.-P.; Hao, X.-J. β-Carboline alkaloids from the leaves of Trigonostemon lii. Bioorg. Med. Chem. Lett. 2012, 22, 2296–2299. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, P.; Kong, F.D.; Wang, J.; Zuo, W.J.; Wang, H.; Dai, H.F.; Mei, W.L. Two new alkaloids from the twigs of Trigonostemon filipes. J. Asian Nat. Prod. Res. 2018, 20, 270–276. [Google Scholar] [CrossRef]
- Machowinski, A.; Krämer, H.J.; Hort, W.; Mayser, P. Pityriacitrin—A potent UV filter produced by Malassezia furfur and its effect on human skin microflora. Mycoses 2006, 49, 388–392. [Google Scholar] [CrossRef]
- Nagao, T.; Adachi, K.; Nishida, F.; Nishishima, M.; Mochida, K. New Ultraviolet-Absorbing Substance Produced by Marine Bacteria and Its Production. JP Patent 11,269,175, 5 October 1999. [Google Scholar]
- Chen, Y.-X.; Xu, M.-Y.; Li, H.-J.; Zeng, K.-J.; Ma, W.-Z.; Tian, G.-B.; Xu, J.; Yang, D.-P.; Lan, W.-J. Diverse secondary metabolites from the marine-derived fungus Dichotomomyces cejpii F31-1. Mar. Drugs 2017, 15, 339. [Google Scholar] [CrossRef] [Green Version]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.-C.; van Soest, R.W. 5-Hydroxytryptamine-derived alkaloids from two marine sponges of the genus Hyrtios. J. Nat. Prod. 2002, 65, 1173–1176. [Google Scholar] [CrossRef]
- Huang, W.; Yi, X.; Feng, J.; Wang, Y.; He, X. Piperidine alkaloids from Alocasia macrorrhiza. Phytochemistry 2017, 143, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Cheng, Y.Y.; Zhang, Y.; Li, S.L.; He, H.P.; Hao, X.J. β-carboline alkaloids from Trigonostemon filipes and Trigonostemon lii. Nat. Prod. Bioprospect. 2012, 2, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Sun, X.; Xu, B.; Bijian, K.; Wan, S.; Li, G.; Alaoui-Jamali, M.; Jiang, T. Total synthesis and bioactivity of the marine alkaloid pityriacitrin and some of its derivatives. Eur. J. Med. Chem. 2011, 46, 6089–6097. [Google Scholar] [CrossRef] [PubMed]
- Liew, L.P.; Fleming, J.M.; Longeon, A.; Mouray, E.; Florent, I.; Bourguet-Kondracki, M.-L.; Copp, B.R. Synthesis of 1-indolyl substituted β-carboline natural products and discovery of antimalarial and cytotoxic activities. Tetrahedron 2014, 70, 4910–4920. [Google Scholar] [CrossRef]
- Mexia, N.; Gaitanis, G.; Velegraki, A.; Soshilov, A.; Denison, M.S.; Magiatis, P. Pityriazepin and other potent AhR ligands isolated from Malassezia furfur yeast. Arch. Biochem. Biophys. 2015, 571, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-h.; Chen, C.; Wang, H.; Ye, W.-c.; Zhou, G.-x. Indole alkaloids from Alocasia macrorrhiza. Chem. Pharm. Bull. 2012, 60, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.D.; da Silva, T.; Aguiar, A.C.C.; Oliva, G.; Guido, R.V.; Yokoyama-Yasunaka, J.K.; Uliana, S.R.; Lopes, L.M. Chemical composition, antiprotozoal and cytotoxic activities of indole alkaloids and benzofuran neolignan of Aristolochia cordigera. Planta Med. 2017, 83, 912–920. [Google Scholar] [CrossRef]
- Sauleau, P.; Martin, M.-T.; Dau, M.-E.T.H.; Youssef, D.T.; Bourguet-Kondracki, M.-L. Hyrtiazepine, an azepino-indole-type alkaloid from the Red Sea marine sponge Hyrtios erectus. J. Nat. Prod. 2006, 69, 1676–1679. [Google Scholar] [CrossRef]
- Diallo, A.O.; Mehri, H.; Iouzalen, L.; Plat, M. Alkaloids from leaves of Alangium bussyanum. Phytochemistry 1995, 40, 975–977. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Carte, B.; Taylor, P.B.; Johnson, R.K.; Faulkner, D.J. Haploscleridamine, a novel tryptamine-derived alkaloid from a aponge of the order Haplosclerida: An inhibitor of Cathepsin K. J. Nat. Prod. 2002, 65, 628–629. [Google Scholar] [CrossRef]
- Wang, K.B.; Li, S.G.; Huang, X.Y.; Li, D.H.; Li, Z.L.; Hua, H.M. (±)-Peharmaline A: A pair of rare β-carboline–vasicinone hybrid alkaloid enantiomers from Peganum harmala. Eur. J. Org. Chem. 2017, 2017, 1876–1879. [Google Scholar] [CrossRef]
- Wang, S.G.; Xia, Z.L.; Xu, R.Q.; Liu, X.J.; Zheng, C.; You, S.L. Construction of chiral tetrahydro-β-carbolines: Asymmetric Pictet–Spengler reaction of indolyl dihydropyridines. Angew. Chem. 2017, 129, 7548–7551. [Google Scholar] [CrossRef]
- Tulyaganov, T.S.; Nazarov, O.M.; Levkovich, M.G.; Abdullaev, N.D. Alkaloids of the Nitraria genus. Komavine and Acetylkomavine. Chem. Nat. Compd. 2001, 37, 61–64. [Google Scholar] [CrossRef]
- Huang, H.; Yao, Y.; He, Z.; Yang, T.; Ma, J.; Tian, X.; Li, Y.; Huang, C.; Chen, X.; Li, W. Antimalarial β-carboline and indolactam alkaloids from Marinactinospora thermotolerans, a deep sea isolate. J. Nat. Prod. 2011, 74, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, Y.; Jin, H.J.; Cui, Y.D.; Zhang, L.J.; Jiang, T. An efficient synthesis method targeted to marine alkaloids marinacarbolines A-D and their antitumor activities. J. Asian Nat. Prod. Res. 2015, 17, 299–305. [Google Scholar] [CrossRef]
- Jaeger, R.J.; Lamshöft, M.; Gottfried, S.; Spiteller, M.; Spiteller, P. HR-MALDI-MS imaging assisted screening of β-carboline alkaloids discovered from Mycena metata. J. Nat. Prod. 2013, 76, 127–134. [Google Scholar] [CrossRef]
- Naveen, B.; Mudiraj, A.; Khamushavalli, G.; Babu, P.P.; Nagarajan, R. Concise total synthesis of water soluble metatacarboline A, C, D, E and F and its anticancer activity. Eur. J. Med. Chem. 2016, 113, 167–178. [Google Scholar] [CrossRef]
- Oku, N.; Matsunaga, S.; Fusetani, N. Shishijimicins A−C, novel enediyne antitumor antibiotics from the ascidian Didemnum proliferum. J. Am. Chem. Soc. 2003, 125, 2044–2045. [Google Scholar] [CrossRef]
- Ohmoto, T.; Koike, K. Chapter 3 Canthin-6-one Alkaloids. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Academic Press: New York, NY, USA, 1990; Volume 36, pp. 135–170. [Google Scholar]
- Kam, T.-S.; Sim, K.-M. Alkaloids from Kopsia griffithii. Phytochemistry 1998, 47, 145–147. [Google Scholar] [CrossRef]
- Wen-sen, C. The isolation and structure of cordatanine from Drymaria cordata (L.) Willd. J. Integr. Plant Biol. 1986, 28, 450–452. [Google Scholar]
- Hsieh, P.-W.; Chang, F.-R.; Lee, K.-H.; Hwang, T.-L.; Chang, S.-M.; Wu, Y.-C. A new anti-HIV alkaloid, drymaritin, and a new C-glycoside flavonoid, diandraflavone, from Drymaria diandra. J. Nat. Prod. 2004, 67, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, I.; Allmendinger, L.; Bracher, F. Revised structure of the alkaloid drymaritin. J. Nat. Prod. 2009, 72, 1908–1910. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Genta-Jouve, G.; Zheng, Y.; Zhang, Q.; Chen, C.; Zhou, Q.; Wang, J.; Zhu, H.; Zhang, Y. Griseofamines A and B: Two Indole-Tetramic Acid Alkaloids with 6/5/6/5 and 6/5/7/5 Ring Systems from Penicillium griseofulvum. Org. Lett. 2018, 20, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, Z. Total synthesis and antibacterial activity evaluation of griseofamine A and 16-epi-griseofamine A. Org. Lett. 2019, 21, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Besslièvre, R.; Cosson, B.P.; Das, B.C.; Husson, H.P. Structure and total synthesis of deplancheine, a novel indoloquinolizidine alkaloid. Tetrahedron Lett. 1980, 21, 63–66. [Google Scholar] [CrossRef]
- Shi, B.B.; Chen, J.; Bao, M.F.; Zeng, Y.; Cai, X.H. Alkaloids isolated from Tabernaemontana bufalina display xanthine oxidase inhibitory activity. Phytochemistry 2019, 166, 112060. [Google Scholar] [CrossRef]
- Bao, M.-F.; Zeng, C.-X.; Liu, Y.-P.; Zhang, B.-J.; Ni, L.; Luo, X.-D.; Cai, X.-H. Indole alkaloids from Hunteria zeylanica. J. Nat. Prod. 2017, 80, 790–797. [Google Scholar] [CrossRef]
- Yan, W.; Ge, H.M.; Wang, G.; Jiang, N.; Mei, Y.N.; Jiang, R.; Li, S.J.; Chen, C.J.; Jiao, R.H.; Xu, Q.; et al. Pictet-Spengler reaction-based biosynthetic machinery in fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 18138–18143. [Google Scholar] [CrossRef] [Green Version]
- Khokhar, S.; Feng, Y.; Campitelli, M.R.; Ekins, M.G.; Hooper, J.N.A.; Beattie, K.D.; Sadowski, M.C.; Nelson, C.C.; Davis, R.A. Isolation, structure determination and cytotoxicity studies of tryptophan alkaloids from an Australian marine sponge Hyrtios sp. Bioorg. Med. Chem. Lett. 2014, 24, 3329–3332. [Google Scholar] [CrossRef]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer activity of fascaplysin against lung cancer cell and small cell lung cancer circulating tumor cell lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [Green Version]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.J.; Kashiwaki, K. Chemical constituents of the fruits of Evodia rutaecarpa. Pharm. Soc. Jpn. 1915, 405, 1293. [Google Scholar]
- Sun, Q.; Xie, L.; Song, J.; Li, X. Evodiamine: A review of its pharmacology, toxicity, pharmacokinetics and preparation researches. J. Ethnopharmacol. 2020, 262, 113164. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.M.A.G.; Court, W.E. Stem bark alkaloids of rauvolfia caffra. J. Ethnopharmacol. 1984, 11, 99–117. [Google Scholar] [CrossRef]
- Majumdar, S.P.; Potier, P.; Poisson, J. The structure of suaveoline, a new alkaloid from Rauwolfia suaveolens S. Moore (apocynaceae). Tetrahedron Lett. 1972, 13, 1563–1566. [Google Scholar] [CrossRef]
- Amer, M.M.A.; Court, W.E. Stem bark alkaloids of Rauwolfia macrophylla. Planta Med. 1980, 40, 8–12. [Google Scholar] [CrossRef]
- Zhan, G.; Miao, R.; Zhang, F.; Hao, X.; Zheng, X.; Zhang, H.; Zhang, X.; Guo, Z. Monoterpene indole alkaloids with diverse skeletons from the stems of Rauvolfia vomitoria and their acetylcholinesterase inhibitory activities. Phytochemistry 2020, 177, 112450. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Tang, C.-P.; Ke, C.-Q.; Ye, Y. Indole alkaloids from leaves and stems of Hunteria zeylanica. Chem. Nat. Compd. 2009, 45, 834–836. [Google Scholar] [CrossRef]
- Schnoes, H.K.; Biemann, K.; Mokry, J.; Kompis, I.; Chatterjee, A.; Ganguli, G. Strictamine. J. Org. Chem. 1966, 31, 1641–1642. [Google Scholar] [CrossRef]
- Ahmad, Y.; Fatima, K.; Atta ur, R.; Occolowitz, J.L.; Solheim, B.A.; Clardy, J.; Garnick, R.L.; Le Quesne, P.W. Structure and absolute configuration of strictamine and strictalamine from Rhazya stricta. Stereochemistry of the Picralima alkaloids. J. Am. Chem. Soc. 1977, 99, 1943–1946. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.-J.; Zhang, D.-B.; Wen, J.; Zhao, X.-W.; Li, Y.; Gao, K. An unusual indole alkaloid with anti-adenovirus and anti-HSV activities from Alstonia scholaris. Tetrahedron Lett. 2014, 55, 1815–1817. [Google Scholar] [CrossRef]
- Wong, S.-P.; Chong, K.-W.; Lim, K.-H.; Lim, S.-H.; Low, Y.-Y.; Kam, T.-S. Arborisidine and arbornamine, two monoterpenoid indole alkaloids with new polycyclic carbon–nitrogen skeletons derived from a common pericine precursor. Org. Lett. 2016, 18, 1618–1621. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, T.A.; Verpoorte, R.; Baerheim Svendsen, A. Alkaloids of Tabernaemontana eglandulosa. Tetrahedron 1984, 40, 737–748. [Google Scholar] [CrossRef]
- Gilani, S.A.; Kikuchi, A.; Shinwari, Z.K.; Khattak, Z.I.; Watanabe, K.N. Phytochemical, pharmacological and ethnobotanical studies of Rhazya stricta Decne. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2007, 21, 301–307. [Google Scholar] [CrossRef]
- Kitajima, M.; Nakano, S.; Kogure, N.; Subhadhirasakul, S.; Takayama, H. New indole alkaloids from Ervatamia cumingiana (Dedicated to Professor Tohru Fukuyama on the occasion of his 70th birthday). Heterocycles Int. J. Rev. Commun. Heterocycl. Chem. 2019, 99, 213–221. [Google Scholar]
- Ndongo, J.T.; Mbing, J.N.; Monteillier, A.; Tala, M.F.; Rütten, M.; Mombers, D.; Cuendet, M.; Pegnyemb, D.E.; Dittrich, B.; Laatsch, H. Carbazole-, aspidofractinine-, and aspidocarpamine-type alkaloids from Pleiocarpa pycnantha. J. Nat. Prod. 2018, 81, 1193–1202. [Google Scholar] [CrossRef]
- Zhang, B.-J.; Teng, X.-F.; Bao, M.-F.; Zhong, X.-H.; Ni, L.; Cai, X.-H. Cytotoxic indole alkaloids from Tabernaemontana officinalis. Phytochemistry 2015, 120, 46–52. [Google Scholar] [CrossRef]
- Subramaniam, G.; Hiraku, O.; Hayashi, M.; Koyano, T.; Komiyama, K.; Kam, T.-S. Biologically active aspidofractinine alkaloids from Kopsia singapurensis. J. Nat. Prod. 2008, 71, 53–57. [Google Scholar] [CrossRef]
- Lim, S.-H.; Sim, K.-M.; Abdullah, Z.; Hiraku, O.; Hayashi, M.; Komiyama, K.; Kam, T.-S. Leuconoxine, kopsinitarine, kopsijasmine, and kopsinone derivatives from Kopsia. J. Nat. Prod. 2007, 70, 1380–1383. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.-Y.; Qin, X.-J.; Wang, B.; Jin, Q.; Liu, Y.-P.; Luo, X.-D. Antibacterial monoterpenoid indole alkaloids from Alstonia scholaris cultivated in temperate zone. Fitoterapia 2015, 105, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Penelle, J.; Tits, M.; Christen, P.; Molgo, J.; Brandt, V.; Frédérich, M.; Angenot, L. Quaternary indole alkaloids from the stem bark of Strychnos guianensis. Phytochemistry 2000, 53, 1057–1066. [Google Scholar] [CrossRef]
- Naaz, H.; Singh, S.; Pandey, V.P.; Singh, P.; Dwivedi, U.N. Anti-cholinergic alkaloids as potential therapeutic agents for Alzheimer’s disease: An in silico approach. Indian J. Biochem. Biophys. 2013, 50, 120–125. [Google Scholar] [PubMed]
- Kitajima, M.; Anbe, M.; Kogure, N.; Wongseripipatana, S.; Takayama, H. Indole alkaloids from Kopsia jasminiflora. Tetrahedron 2014, 70, 9099–9106. [Google Scholar] [CrossRef]
- Kam, T.-S.; Arasu, L.; Yoganathan, K. Alkaloids from Kopsia pauciflora. Phytochemistry 1996, 43, 1385–1387. [Google Scholar] [CrossRef]
- Walser, A.; Djerassi, C. Alkaloid-Studien LII. Die Alkaloide aus Vallesia dichotoma. Helv. Chim. Acta 1965, 48, 391–404. [Google Scholar] [CrossRef]
- Renner, U. Alkaloide aus Schizozygia Caffaeoides. 3. Strukturelle Beziehungen zwischen Schizozygin und einigen Nebenalkaloiden. Lloydia 1964, 27, 406–415. [Google Scholar]
- Wang, K.-B.; Di, Y.-T.; Bao, Y.; Yuan, C.-M.; Chen, G.; Li, D.-H.; Bai, J.; He, H.-P.; Hao, X.-J.; Pei, Y.-H. Peganumine A, a β-carboline dimer with a new octacyclic scaffold from Peganum harmala. Org. Lett. 2014, 16, 4028–4031. [Google Scholar] [CrossRef]
- Neuss, N.; Boaz, H.E.; Forbes, J. Rauwolfia Serpentina alkaloids. I. Structure of reserpine. J. Am. Chem. Soc. 1954, 76, 2463–2467. [Google Scholar] [CrossRef]
- Tsioufis, C.; Thomopoulos, C. Combination drug treatment in hypertension. Pharmacol. Res. 2017, 125, 266–271. [Google Scholar] [CrossRef]
- De Silva, K.T.D.; King, D.; Smith, G.N. 5α-Carboxystrictosidine. J. Chem. Soc. D Chem. Commun. 1971, 16, 908–909. [Google Scholar] [CrossRef]
- Ferrari, F.; Messana, I.; Botta, B.; de Mello, J.F. Constituents of Guettarda platypoda. J. Nat. Prod. 1986, 49, 1150–1151. [Google Scholar] [CrossRef]
- Kitajima, M.; Hashimoto, K.-i.; Yokoya, M.; Takayama, H.; Aimi, N.; Sakai, S.-i. A New gluco indole alkaloid, 3,4-dehydro-5-carboxystrictosidine, from Preruvian Una de Gato (Uncaria tomentosa). Chem. Pharm. Bull. 2000, 48, 1410–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onozawa, T.; Kitajima, M.; Kogure, N.; Peerakam, N.; Santiarworn, D.; Takayama, H. A Cyclopeptide and a tetrahydroisoquinoline ílkaloid from Ophiorrhiza nutans. J. Nat. Prod. 2017, 80, 2156–2160. [Google Scholar] [CrossRef]
- Brown, R.T.; Charalambides, A.A. Adina alkaloids: The structure of rubenine. J. Chem. Soc. Chem. Commun. 1973, 20, 765–766. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Xiang, J.-C.; Cheng, Y.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. Direct biomimetic synthesis of β-carboline alkaloids from two amino acids. J. Org. Chem. 2018, 83, 12247–12254. [Google Scholar] [CrossRef]
- Lood, C.; Nieger, M.; Koskinen, A. Enantiospecific gram scale synthesis of (S)-eleagnine. Tetrahedron 2015, 71, 5019–5024. [Google Scholar] [CrossRef]
- Ellman, J.A.; Owens, T.D.; Tang, T.P. N-tert-butanesulfinyl imines: Versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 2002, 35, 984–995. [Google Scholar] [CrossRef]
- Reddy, N.S.S.; Babu, R.A.; Reddy, B.V.S. Asymmetric synthesis of tetrahydro-β-carboline alkaloids employing Ellman’s chiral auxiliary. Synthesis 2016, 48, 1079–1086. [Google Scholar] [CrossRef]
- Van Maarseveen, J.H.; Hermkens, P.H.; De Clercq, E.; Balzarini, J.; Scheeren, H.W.; Kruse, C.G. Antiviral and antitumor structure-activity relationship studies on tetracyclic eudistomines. J. Med. Chem. 1992, 35, 3223–3230. [Google Scholar] [CrossRef]
- Roggero, C.M.; Giulietti, J.M.; Mulcahy, S.P. Efficient synthesis of eudistomin U and evaluation of its cytotoxicity. Bioorg. Med. Chem. Lett. 2014, 24, 3549–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodina, A.A.; OV, S. Eudistomin U, isoeudistomin U, and related indole compounds: Synthesis and biological activity. Heterocycles 2018, 96, 1171–1196. [Google Scholar] [CrossRef]
- Kamal, A.; Sathish, M.; Prasanthi, A.V.G.; Chetna, J.; Tangella, Y.; Srinivasulu, V.; Shankaraiah, N.; Alarifi, A. An efficient one-pot decarboxylative aromatization of tetrahydro-β-carbolines by using N-chlorosuccinimide: Total synthesis of norharmane, harmane and eudistomins. RSC Adv. 2015, 5, 90121–90126. [Google Scholar] [CrossRef]
- Davis, R.A.; Carroll, A.R.; Quinn, R.J. Eudistomin V, a New β-carboline from the australian ascidian Pseudodistoma aureum. J. Nat. Prod. 1998, 61, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Sen, S. N-Bromo-succinimide promoted synthesis of β-carbolines and 3,4-dihydro-β-carbolines from tetrahydro-β-carbolines. Tetrahedron Lett. 2016, 57, 1040–1043. [Google Scholar] [CrossRef]
- Kamal, A.; Tangella, Y.; Manasa, K.L.; Sathish, M.; Srinivasulu, V.; Chetna, J.; Alarifi, A. PhI(OAc)2-mediated one-pot oxidative decarboxylation and aromatization of tetrahydro-β-carbolines: Synthesis of norharmane, harmane, eudistomin U and eudistomin I. Org. Biomol. Chem. 2015, 13, 8652–8662. [Google Scholar] [CrossRef] [PubMed]
- Pakhare, D.S.; Kusurkar, R.S. Synthesis of tetrahydro-β-carbolines, β-carbolines, and natural products, (±)-harmicine, eudistomin U and canthine by reductive Pictet–Spengler cyclization. Tetrahedron Lett. 2015, 56, 6012–6015. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Földesi, T.; Grün, A.; Volk, B.; Keglevich, G.; Milen, M. Synthetic study on the T3P®-promoted one-pot preparation of 1-substituted-3,4-dihydro-β-carbolines by the reaction of tryptamine with carboxylic acids. Tetrahedron Lett. 2016, 57, 1953–1957. [Google Scholar] [CrossRef]
- Gaikwad, S.; Kamble, D.; Lokhande, P. Iodine-catalyzed chemoselective dehydrogenation and aromatization of tetrahydro-β-carbolines: A short synthesis of kumujian-C, eudistomin-U, norharmane, harmane harmalan and isoeudistomine M. Tetrahedron Lett. 2018, 59, 2387–2392. [Google Scholar] [CrossRef]
- Szabó, T.; Dancso, A.; Volk, B.; Milen, M. First total synthesis of beta-carboline alkaloid trigonostemine G and its derivatives. Nat. Prod. Res. 2021, 35, 72–79. [Google Scholar] [CrossRef]
- Szabó, T.; Hazai, V.; Volk, B.; Simig, G.; Milen, M. First total synthesis of the β-carboline alkaloids trigonostemine A, trigonostemine B and a new synthesis of pityriacitrin and hyrtiosulawesine. Tetrahedron Lett. 2019, 60, 1471–1475. [Google Scholar] [CrossRef]
- Ambule, M.D.; Tripathi, S.; Ghoshal, A.; Srivastava, A.K. IBX-mediated oxidative addition of isocyanides to cyclic secondary amines: Total syntheses of alangiobussine and alangiobussinine. Chem. Commun. 2019, 55, 10872–10875. [Google Scholar] [CrossRef] [PubMed]
- Singha Roy, M.; Meng, X.; Koda, K.; Rasapalli, S.; Gout, D.; Lovely, C.J. Total synthesis of (−)-haploscleridamine. Tetrahedron Lett. 2019, 60, 979–982. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Shingare, R.D.; Dandela, R.; Reddy, D.S. Total synthesis of an anticancer natural product (±)-peharmaline A and its analogues. Eur. J. Org. Chem. 2018, 2018, 6453–6456. [Google Scholar] [CrossRef]
- Wang, Z.; Niu, J.; Zeng, H.; Li, C.-J. Construction of spirocyclic tetrahydro-β-carbolines via cross-annulation of phenols with tryptamines in water. Org. Lett. 2019, 21, 7033–7037. [Google Scholar] [CrossRef]
- Meng, T.-Z.; Zheng, J.; Trieu, T.H.; Zheng, B.; Wu, J.-J.; Zhang, Y.; Shi, X.-X. CuBr2-Catalyzed mild oxidation of 3,4-dihydro-β-carbolines and application in total synthesis of 6-hydroxymetatacarboline D. ACS Omega 2018, 3, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.; Lu, Z.; Li, R.; Woods, J.R.; Sohn, T.-i. Total synthesis of shishijimicin A. J. Am. Chem. Soc. 2015, 137, 8716–8719. [Google Scholar] [CrossRef]
- Schott, Y.; Decker, M.; Rommelspacher, H.; Lehmann, J. 6-Hydroxy- and 6-methoxy-β-carbolines as acetyl- and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5840–5843. [Google Scholar] [CrossRef]
- Cogan, D.A.; Liu, G.; Ellman, J. Asymmetric synthesis of chiral amines by highly diastereoselective 1,2-additions of organometallic reagents to N-tert-butanesulfinyl imines. Tetrahedron 1999, 55, 8883–8904. [Google Scholar] [CrossRef]
- Davis, F.A.; Gaddiraju, N.V.; Theddu, N.; Hummel, J.R.; Kondaveeti, S.K.; Zdilla, M.J. Enantioselective synthesis of cocaine C-1 analogues using sulfinimines (N-sulfinyl imines). J. Org. Chem. 2012, 77, 2345–2359. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Kitajima, M.; Kogure, N.; Takayama, H. A divergent approach for the total syntheses of cernuane-type and quinolizidine-type Lycopodium alkaloids. Tetrahedron 2009, 65, 1608–1617. [Google Scholar] [CrossRef]
- Fandrick, D.R.; Hart, C.A.; Okafor, I.S.; Mercadante, M.A.; Sanyal, S.; Masters, J.T.; Sarvestani, M.; Fandrick, K.R.; Stockdill, J.L.; Grinberg, N. Copper-catalyzed asymmetric propargylation of cyclic aldimines. Org. Lett. 2016, 18, 6192–6195. [Google Scholar] [CrossRef] [PubMed]
- Nalikezhathu, A.; Cherepakhin, V.; Williams, T.J. Ruthenium Catalyzed Tandem Pictet–Spengler Reaction. Org. Lett. 2020, 22, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Shelar, S.V.; Argade, N.P. Total synthesis of bioactive canthine alkaloid cordatanine comprising in situ double oxidative aromatization of tetrahydrocarbazole. ACS Omega 2017, 2, 3945–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.W.; Liao, Y.-R.; Hwang, T.-L.; Shieh, P.-C.; Lee, K.-H.; Hung, H.-Y.; Wu, T.-S. Total synthesis of cordatanine, structural reassignment of drymaritin, and anti-inflammatory activity of synthetic precursors. Bioorg. Med. Chem. Lett. 2015, 25, 3822–3824. [Google Scholar] [CrossRef]
- Zheng, Y.; Wei, K.; Yang, Y.-R. Total synthesis of (−)-geissoschizol through Ir-catalyzed allylic amidation as the key step. Org. Lett. 2017, 19, 6460–6462. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xu, Z.; Tan, R.; Lei, X. Divergent total synthesis of chaetoglines C to F. J. Org. Chem. 2019, 84, 8766–8770. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Xiang, J.-C.; Wang, M.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. One-Pot Total Synthesis of evodiamine and its analogues through a continuous biscyclization reaction. Org. Lett. 2018, 20, 6380–6383. [Google Scholar] [CrossRef]
- Zhao, Z.; Wei, H.; Xiao, K.; Cheng, B.; Zhai, H.; Li, Y. Facile Synthesis of pyridines from propargyl amines: Concise total synthesis of suaveoline alkaloids. Angew. Chem. 2019, 131, 1160–1164. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Bi, Y.-Z.; Yu, F.-X.; Menzia, G.; Cook, J. Stereospecificity in the Pictet–Spengler reaction. Enantiospecific synthesis of (6S,10S)--(–)-5-methyl-9-oxo-12-benzyl-6,7,8,9,10,11-hexahydro-6,10-imino-5H-cyclooct[b]indole, a template for preparation of macroline/sarpagine alkaloids. Heterocycles 1992, 34, 517–547. [Google Scholar]
- Fu, X.; Cook, J.M. General approach for the synthesis of ajmaline-related alkaloids. Enantiospecific total synthesis of (-)-suaveoline, (-)-raumacline, and (-)-Nb-methylraumacline. J. Org. Chem. 1993, 58, 661–672. [Google Scholar] [CrossRef]
- Smith, M.W.; Zhou, Z.; Gao, A.X.; Shimbayashi, T.; Snyder, S.A. A 7-step formal asymmetric total synthesis of strictamine via an asymmetric propargylation and metal-mediated cyclization. Org. Lett. 2017, 19, 1004–1007. [Google Scholar] [CrossRef]
- Ren, W.; Wang, Q.; Zhu, J. Total synthesis of (±)-strictamine. Angew. Chem. Int. Ed. 2016, 55, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, D.; Ohara, A.; Chiba, H.; Kumagai, H.; Oishi, S.; Fujii, N.; Ohno, H. Formal total synthesis of (±)-strictamine based on a gold-catalyzed cyclization. Org. Lett. 2016, 18, 1670–1673. [Google Scholar] [CrossRef] [Green Version]
- Kolundžić, F.; Murali, A.; Pérez-Galán, P.; Bauer, J.O.; Strohmann, C.; Kumar, K.; Waldmann, H. A cyclization–rearrangement cascade for the synthesis of structurally complex chiral gold(I)–aminocarbene complexes. Angew. Chem. Int. Ed. 2014, 53, 8122–8126. [Google Scholar] [CrossRef]
- Zheng, Y.; Yue, B.-B.; Wei, K.; Yang, Y.-R. Short synthesis of the monoterpene indole alkaloid (±)-arbornamine. J. Org. Chem. 2018, 83, 4867–4870. [Google Scholar] [CrossRef]
- Szántay, C.N. The Chemistry of Heterocyclic Compounds; Wiley: Chichester, UK, 1994; Volume 25, Chapter 9. [Google Scholar]
- Vas, Á.; Gulyás, B. Eburnamine derivatives and the brain. Med. Res. Rev. 2005, 25, 737–757. [Google Scholar] [CrossRef]
- Massiot, G.; Oliveira, F.; Lévy, J. Synthesis in the indole series. 9. Total synthesis of the pseudovincamones. Bull. Soc. Chim. Fr. Partie 2 1982, 185–190. [Google Scholar]
- Smith, M.W.; Ferreira, J.; Hunter, R.; Venter, G.A.; Su, H. Synthesis of (+)-tacamonine via stereoselective radical cyclization. Org. Lett. 2019, 21, 8740–8745. [Google Scholar] [CrossRef]
- Jarret, M.; Tap, A.; Kouklovsky, C.; Poupon, E.; Evanno, L.; Vincent, G. Bioinspired oxidative cyclization of the geissoschizine skeleton for the total synthesis of (−)-17-nor-excelsinidine. Angew. Chem. Int. Ed. 2018, 57, 12294–12298. [Google Scholar] [CrossRef] [PubMed]
- Maxime, J.; Victor, T.; Aurélien, T.; Jean-Francois, G.; Cyrille, K.; Erwan, P.; Guillaume, V.; Laurent, E. Bioinspired oxidative cyclization of the geissoschizine skeleton for the enantioselective total synthesis of mavacuran alkaloids. Angew. Chem. Int. Ed. 2019, 58, 9861–9865. [Google Scholar] [CrossRef]
- Sato, K.; Kogure, N.; Kitajima, M.; Takayama, H. Total syntheses of pleiocarpamine, normavacurine, and C-mavacurine. Org. Lett. 2019, 21, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Bai, Y.; Bai, W.-J.; Schultz, J.E. Enantioselective divergent synthesis of C19-oxo eburnane alkaloids via palladium-catalyzed asymmetric allylic alkylation of an N-alkyl-α,β-unsaturated lactam. J. Am. Chem. Soc. 2019, 141, 4811–4814. [Google Scholar] [CrossRef]
- Baran, P.S.; Guerrero, C.A.; Corey, E. Short, enantioselective total synthesis of okaramine N. J. Am. Chem. Soc. 2003, 125, 5628–5629. [Google Scholar] [CrossRef]
- White, K.L.; Mewald, M.; Movassaghi, M. Direct observation of intermediates involved in the interruption of the Bischler–Napieralski reaction. J. Org. Chem. 2015, 80, 7403–7411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Anderson, J.C. A divergent synthetic route to the vallesamidine and schizozygine alkaloids: Total synthesis of (+)-vallesamidine and (+)-14,15-dehydrostrempeliopine. Angew. Chem. Int. Ed. 2019, 58, 18040–18045. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D. Ni(II)−Bis[(R,R)-N,N‘-dibenzylcyclohexane-1,2-diamine]Br2 catalyzed enantioselective Michael additions of 1,3-dicarbonyl compounds to conjugated nitroalkenes. J. Am. Chem. Soc. 2005, 127, 9958–9959. [Google Scholar] [CrossRef]
- Piemontesi, C.; Wang, Q.; Zhu, J. Enantioselective synthesis of (+)-peganumine A. J. Am. Chem. Soc. 2016, 138, 11148–11151. [Google Scholar] [CrossRef]
- Woodward, R.; Bader, F.; Bickel, H.; Frey, A.; Kierstead, R. The total synthesis of reserpine. Tetrahedron 1958, 2, 1–57. [Google Scholar] [CrossRef]
- Khan, Z.A.; Shahzad, S.A.; Anjum, A.; Bale, A.T.; Naqvi, S.A.R. Synthetic approaches toward the reserpine. Synth. Commun. 2018, 48, 1128–1147. [Google Scholar] [CrossRef]
- Park, J.; Chen, D.Y. A desymmetrization-based total synthesis of reserpine. Angew. Chem. Int. Ed. Engl. 2018, 57, 16152–16156. [Google Scholar] [CrossRef] [PubMed]
- Rakumitsu, K.; Sakamoto, J.; Ishikawa, H. Total Syntheses of (−)-Secologanin,(−)-5-Carboxystrictosidine, and (−)-Rubenine. Chem. A Eur. J. 2019, 25, 8996–9000. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Fujiwara, R.; Kasai, Y.; Kitajima, M.; Aimi, N. First asymmetric total synthesis of Us-7 and -8, novel D-seco corynanthe-type oxindole alkaloids from Uncaria a ttenuata: Structure revision of Us-7 and determination of absolute stereochemistry. Org. Lett. 2003, 5, 2967–2970. [Google Scholar] [CrossRef]
- Tietze, L.F.; Meier, H.; Nutt, H. Inter- and intramolecular hetero Diels-Alder reactions, 28. Synthesis of (±)-secologanin aglucone O-ethyl ether and derivatives by tandem Knoevenagel hetero Diels-Alder reaction. Liebigs Ann. Chem. 1990, 1990, 253–260. [Google Scholar] [CrossRef]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R. Exceptional stereochemical control in the Pictet–Spengler reaction. Tetrahedron Lett. 1987, 28, 5177–5180. [Google Scholar] [CrossRef]
Figure 1.
Structure of carboline skeleton and drugs containing a β-carboline framework.

Scheme 1.
One-pot synthesis of natural product norharman (1) and further β-carboline derivatives.

Scheme 2.
Enantioselective synthesis of (S)-eleagnine (2).

Scheme 3.
Synthesis of (−)-tetrahydroharman (2), (−)-komaroidine (3), N-(+)-methyltetrahydroharman (4), (+)-1-ethyl-9-methyltetrahydro-β-carboline (5) and (+)-N-acetylkomaroidine (6).
Scheme 3.
Synthesis of (−)-tetrahydroharman (2), (−)-komaroidine (3), N-(+)-methyltetrahydroharman (4), (+)-1-ethyl-9-methyltetrahydro-β-carboline (5) and (+)-N-acetylkomaroidine (6).

Scheme 4.
Synthesis of stolonine C (7).

Scheme 5.
Kamal’s improved synthesis of eudistomins I (8), N (9), T (10) and U (11).

Scheme 6.
Kamal’s synthesis of eudistomins U (10) and I (8).

Scheme 7.
Synthesis of eudistomin U (10).

Scheme 8.
Ábrányi-Balogh’s synthesis of isoeudistomin U (12).

Scheme 9.
Synthesis of harmalan (13), isoeudistomin M (14), norharman (1), harman (15), eudistomin U (10) and kumujian C (16) via iodine-mediated dehydrogenation.
Scheme 9.
Synthesis of harmalan (13), isoeudistomin M (14), norharman (1), harman (15), eudistomin U (10) and kumujian C (16) via iodine-mediated dehydrogenation.

Scheme 10.
Total synthesis of trigonostemines A (17), B (18) and G (19) and a novel synthetic route for the preparation of pityriacitrin (20) and hyrtiosulawesine (21).
Scheme 10.
Total synthesis of trigonostemines A (17), B (18) and G (19) and a novel synthetic route for the preparation of pityriacitrin (20) and hyrtiosulawesine (21).

Scheme 11.
Synthesis of alangiobussine (22) and alangiobussinine (23).

Scheme 12.
Synthesis of haploscleridamine (24).

Scheme 13.
Synthesis of (±)-peharmaline A (25).

Scheme 14.
Pd-catalyzed cross-annulation of phenols (170) and tryptamines (118).

Scheme 15.
Synthesis of alkaloids marinacarboline A–D (27–30).

Scheme 16.
Total synthesis of metatacarbolines A, C, D, E and F (31–35).

Scheme 17.
Synthesis of 6-hydroxymetatacarboline D (36).

Scheme 18.
Total synthesis of shishijimicin A (37)—Part 1: Synthesis of β-carboline framework 191.

Scheme 19.
Total synthesis of shishijimicin A (37)—Part 2: Synthesis of disaccharide aldehyde 198.

Scheme 20.
Total synthesis of shishijimicin A (31)—Part 3: Synthesis of enediyne thioacetate precursor 212.
Scheme 20.
Total synthesis of shishijimicin A (31)—Part 3: Synthesis of enediyne thioacetate precursor 212.

Scheme 21.
Total synthesis of shishijimicin A (37).

Scheme 22.
Synthesis of canthine (32) and harmicine (33) via reductive Pictet–Spengler cyclization.

Scheme 23.
Synthesis of (−)-harmicine ((−)-39).

Scheme 24.
Synthesis of (−)-harmicine ((−)-39).

Scheme 25.
One-step synthesis of racemic harmicine (39).

Scheme 26.
Five-step synthesis of cordatanine (40).

Scheme 27.
Alternate synthesis of cordatanine (40).

Scheme 28.
Total synthesis of griseofamine A (41).

Scheme 29.
Total synthesis of (+)-deplancheine (42).

Scheme 30.
Synthesis of (−)-geissoschizol (43).

Scheme 31.
Synthesis of chaetoglines E (44) and F (45).

Scheme 32.
First synthesis of 6-oxofascaplysin (46) and a new synthetic route for the preparation of fascaplysin (47).
Scheme 32.
First synthesis of 6-oxofascaplysin (46) and a new synthetic route for the preparation of fascaplysin (47).

Scheme 33.
One-step synthesis of evodiamine (48).

Scheme 34.
Synthesis of norsuaveoline (49), suaveoline (50) and macrophylline (51).

Scheme 35.
Synthesis of (S)-(−)-decarbomethoxy-dihydrogambirtannine (52) and formal synthesis of (+)-strictamine (53).
Scheme 35.
Synthesis of (S)-(−)-decarbomethoxy-dihydrogambirtannine (52) and formal synthesis of (+)-strictamine (53).

Scheme 36.
Formal total synthesis of (±)-strictamine (53).

Scheme 37.
Synthesis of (±)-arbornamine (54).

Scheme 38.
Synthesis of (+)-tacamonine (55).

Scheme 39.
Total synthesis of (−)-17-nor-excelisidine (56) and (+)-geissoschizine (57) by applying direct oxidative cyclization.
Scheme 39.
Total synthesis of (−)-17-nor-excelisidine (56) and (+)-geissoschizine (57) by applying direct oxidative cyclization.

Scheme 40.
Synthesis of (+)-16-epi-pleiocarpamine (58).

Scheme 41.
Synthesis of (+)-16-epi-pleiocarpamine (58), (+)-taberdivarine H (59) and (+)-16-hydroxymethyl-pleiocarpamine (60).
Scheme 41.
Synthesis of (+)-16-epi-pleiocarpamine (58), (+)-taberdivarine H (59) and (+)-16-hydroxymethyl-pleiocarpamine (60).

Scheme 42.
Total synthesis of C-mavacurine-type alkaloids (61–63).

Scheme 43.
Synthesis of eburnane alkaloids (64–67).

Scheme 44.
Synthesis of (+)-vallesamidine (68) and (+)-14,15-dehydrostrempeliopine (69) via key intermediate 359.
Scheme 44.
Synthesis of (+)-vallesamidine (68) and (+)-14,15-dehydrostrempeliopine (69) via key intermediate 359.

Scheme 45.
First total synthesis of (+)-peganumine A (70).

Scheme 46.
Synthesis of reserpine (71).

Scheme 47.
Total synthesis of (−)-5-carboxystrictosidine (72).

Scheme 48.
Total synthesis of (−)-rubenine (73).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
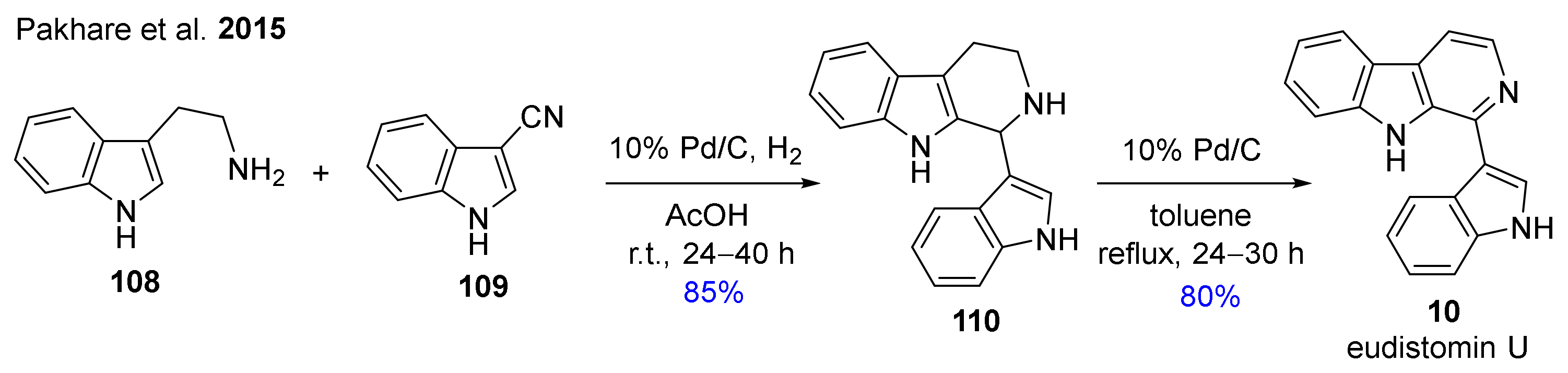{kind=link}
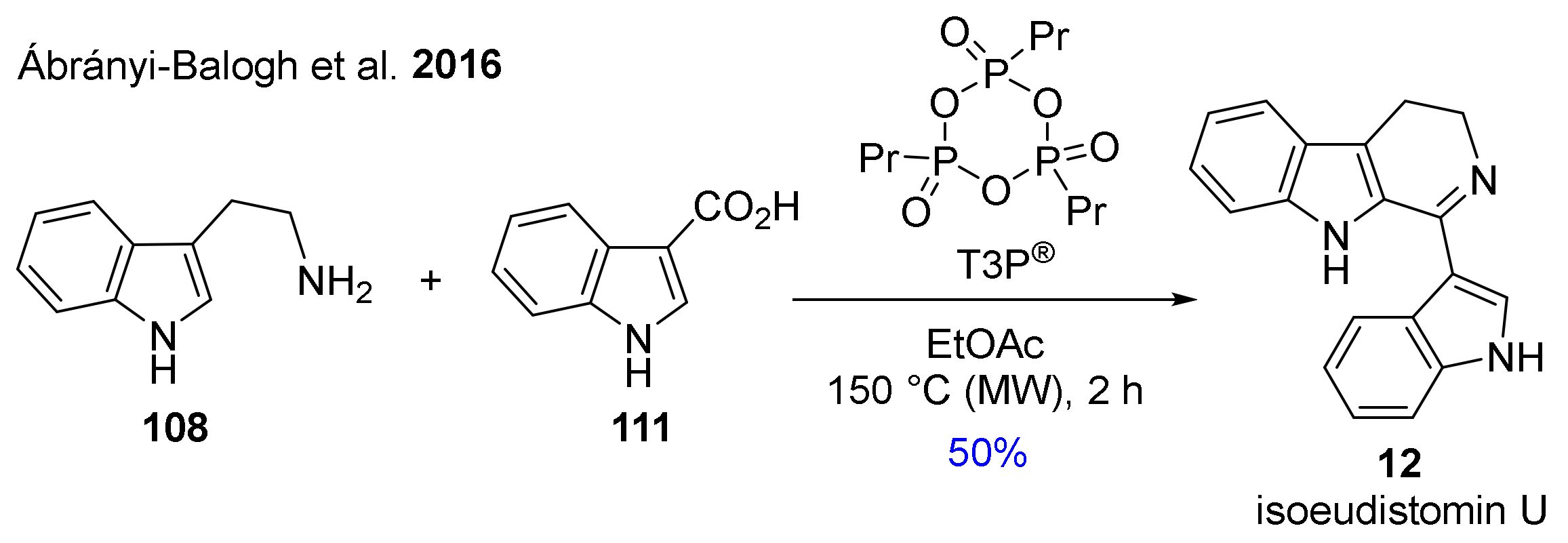{kind=link}
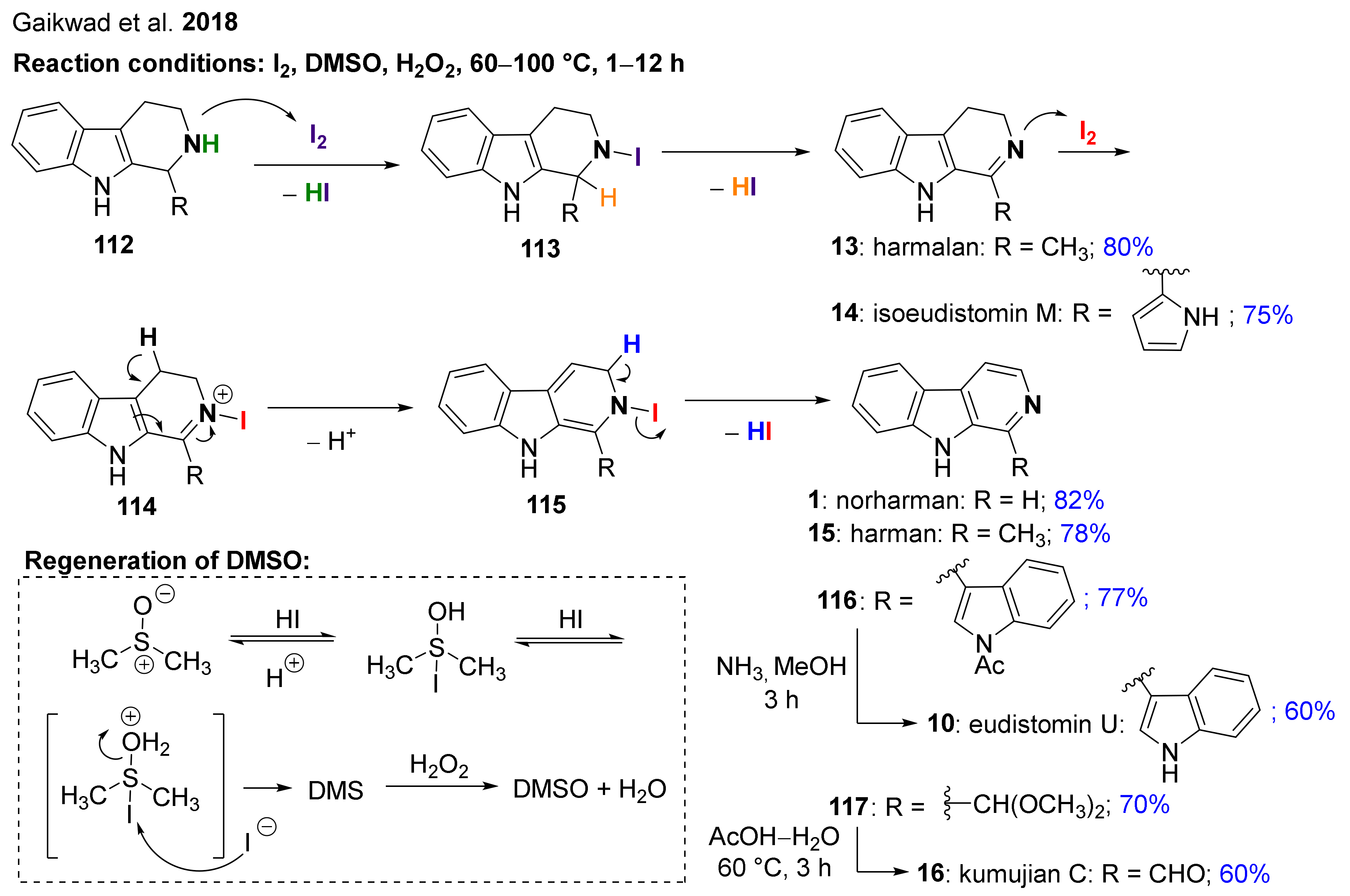{kind=link}
{kind=link}
{kind=link}
{kind=link}
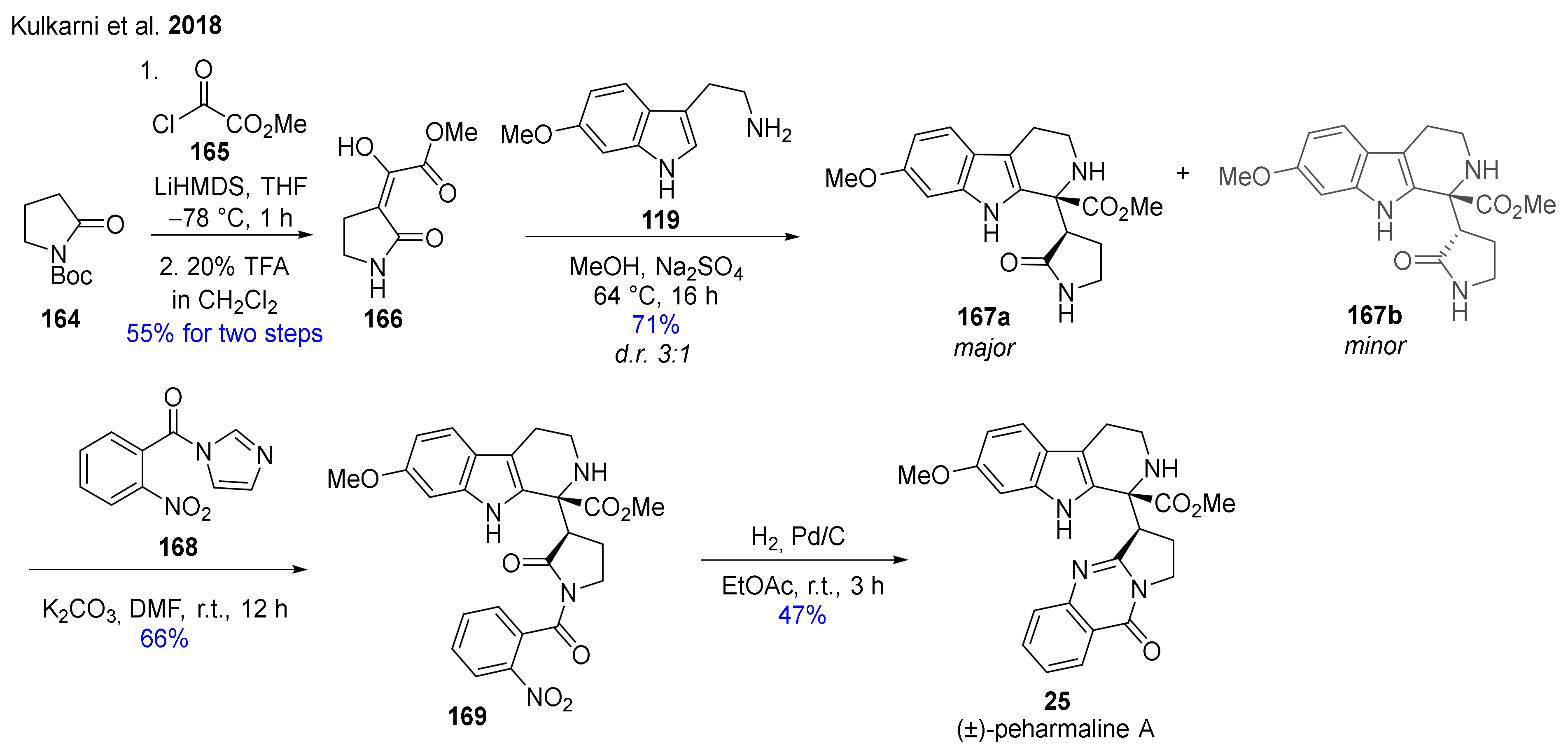{kind=link}
{kind=link}
{kind=link}
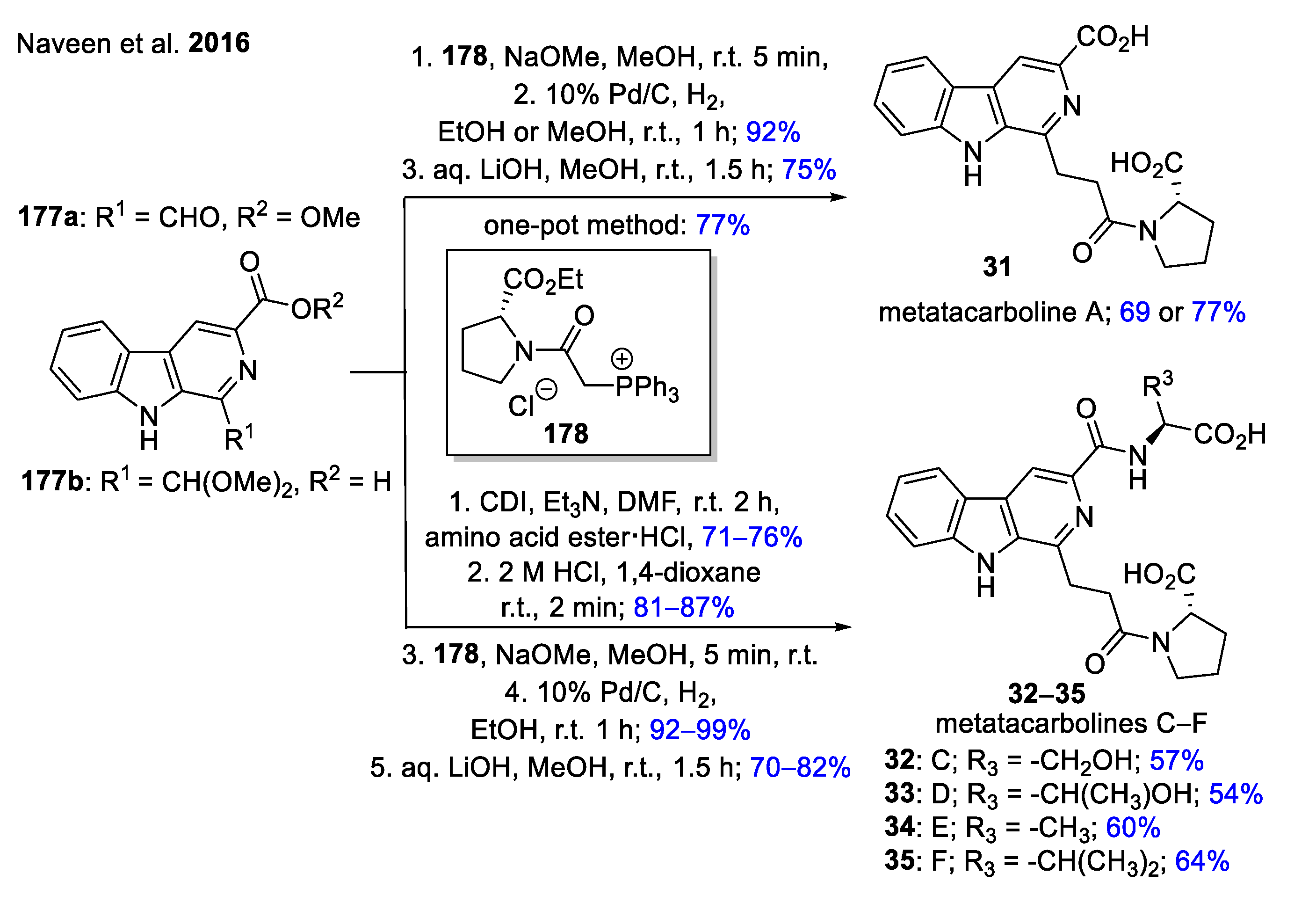{kind=link}
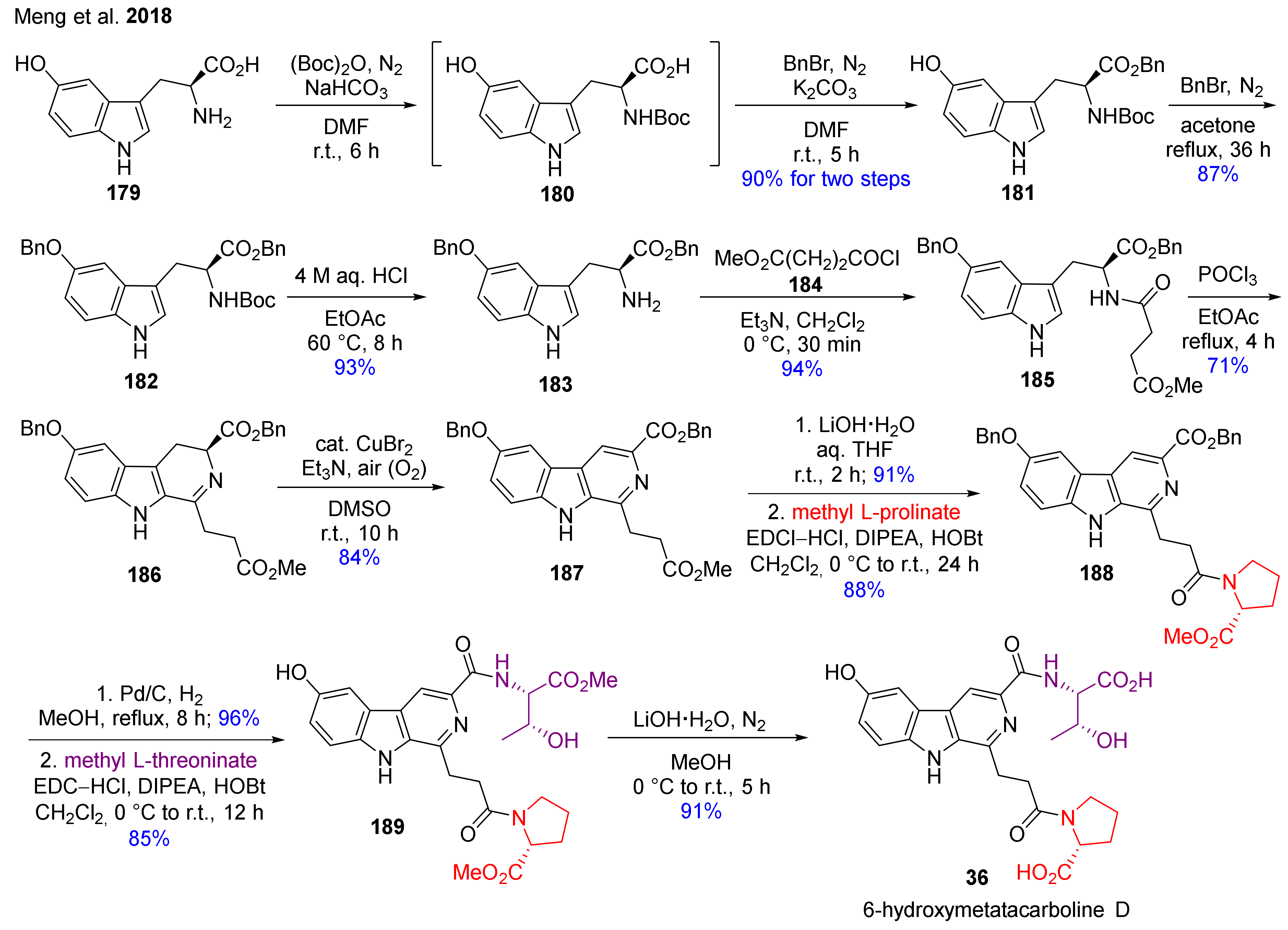{kind=link}
{kind=link}
{kind=link}
{kind=link}
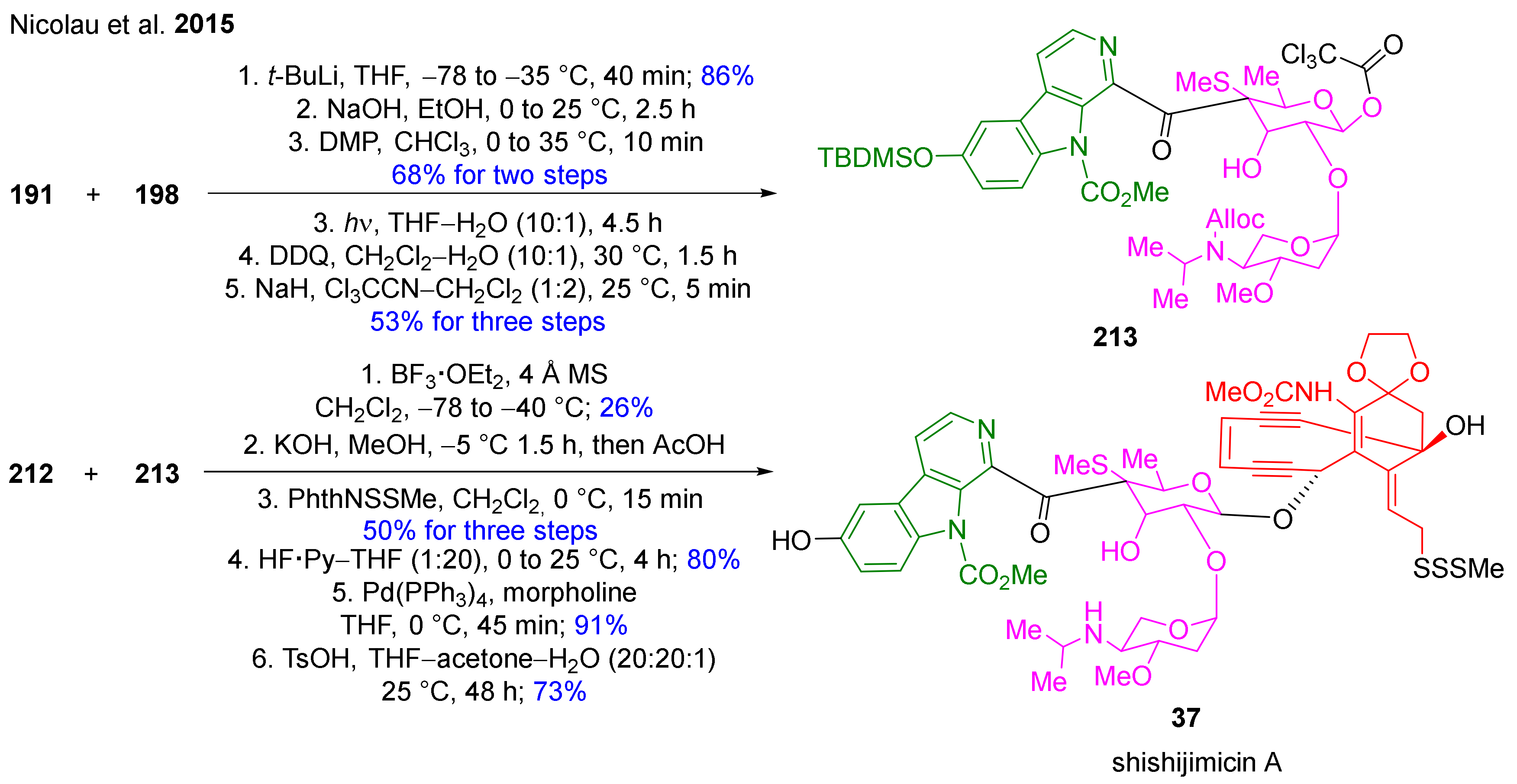{kind=link}
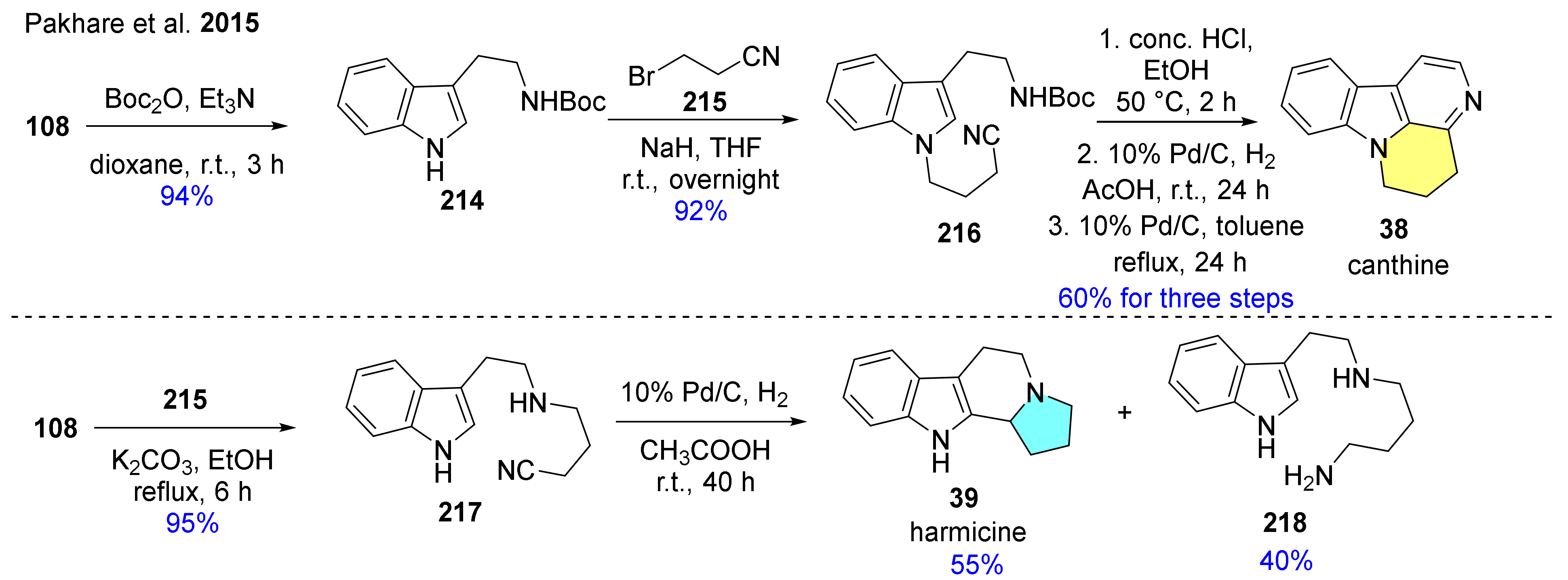{kind=link}
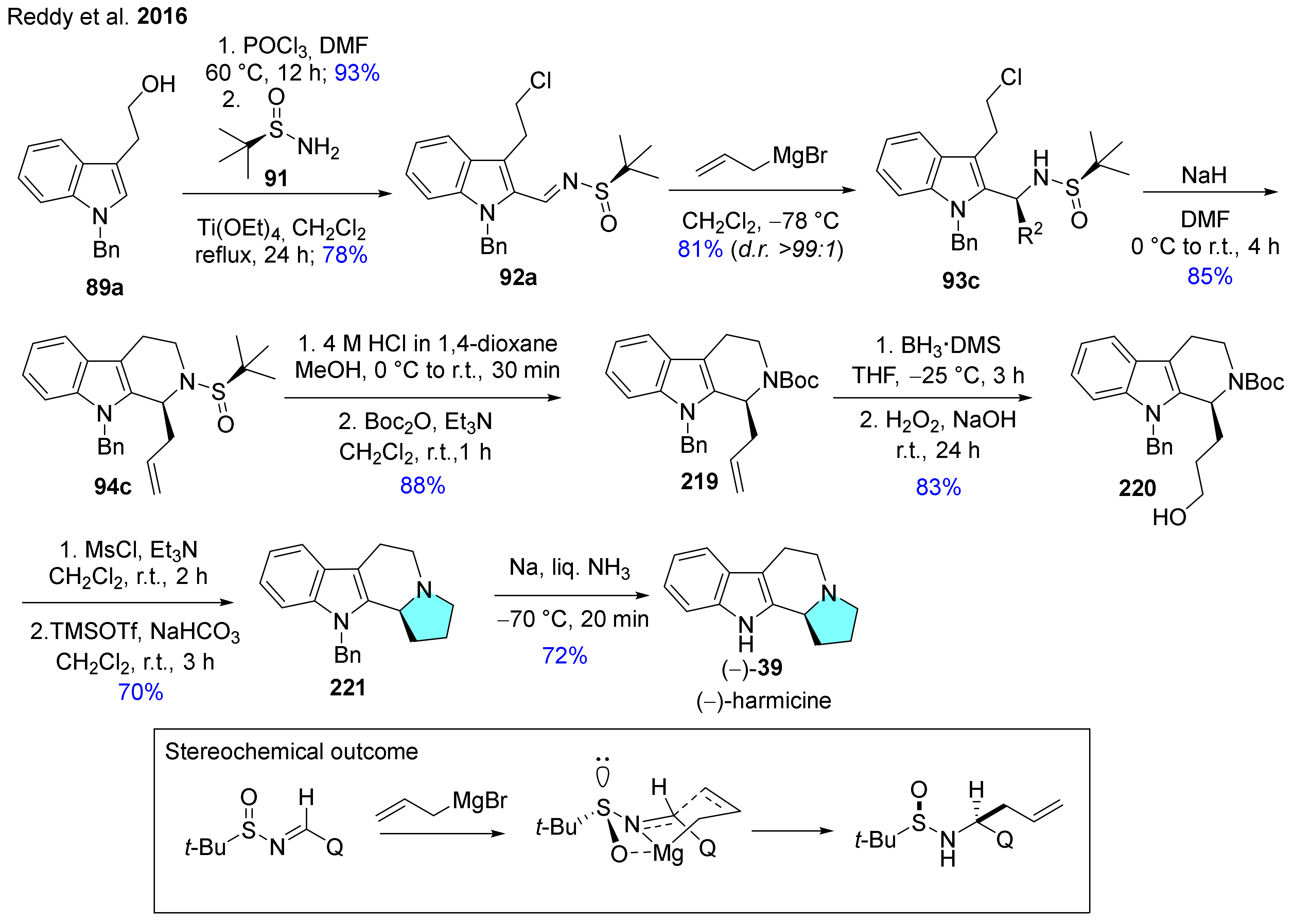{kind=link}
{kind=link}
{kind=link}
{kind=link}
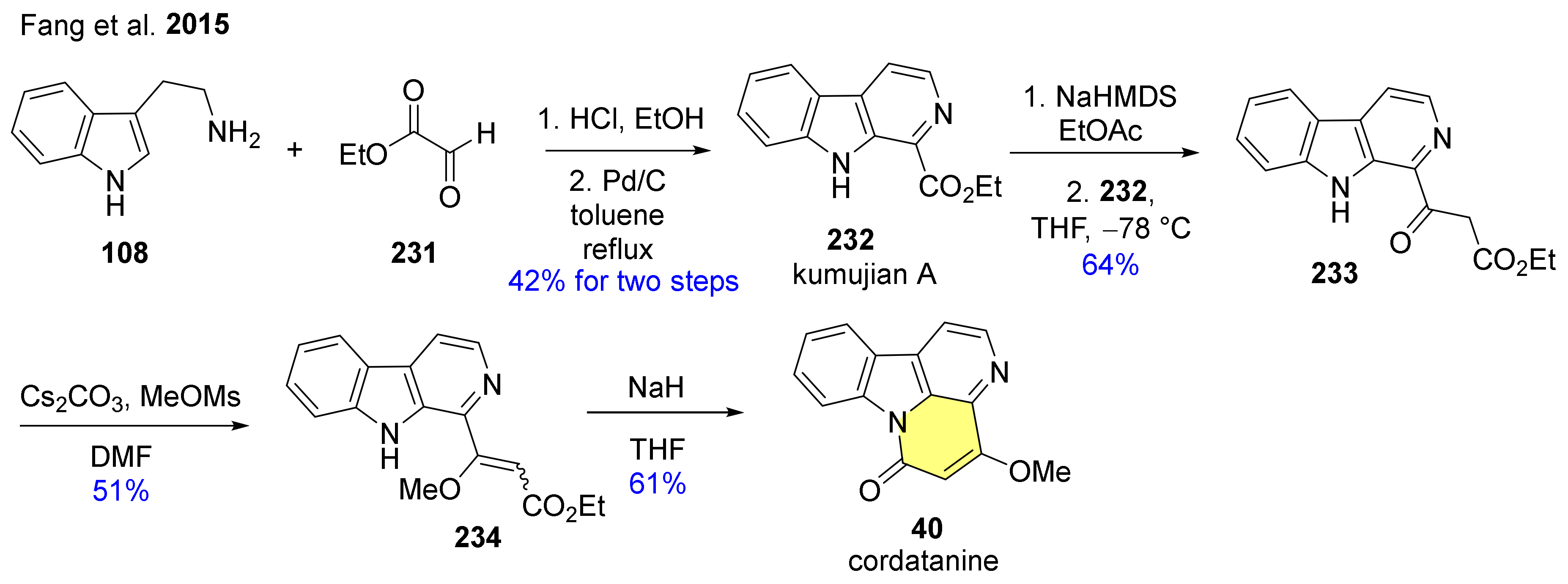{kind=link}
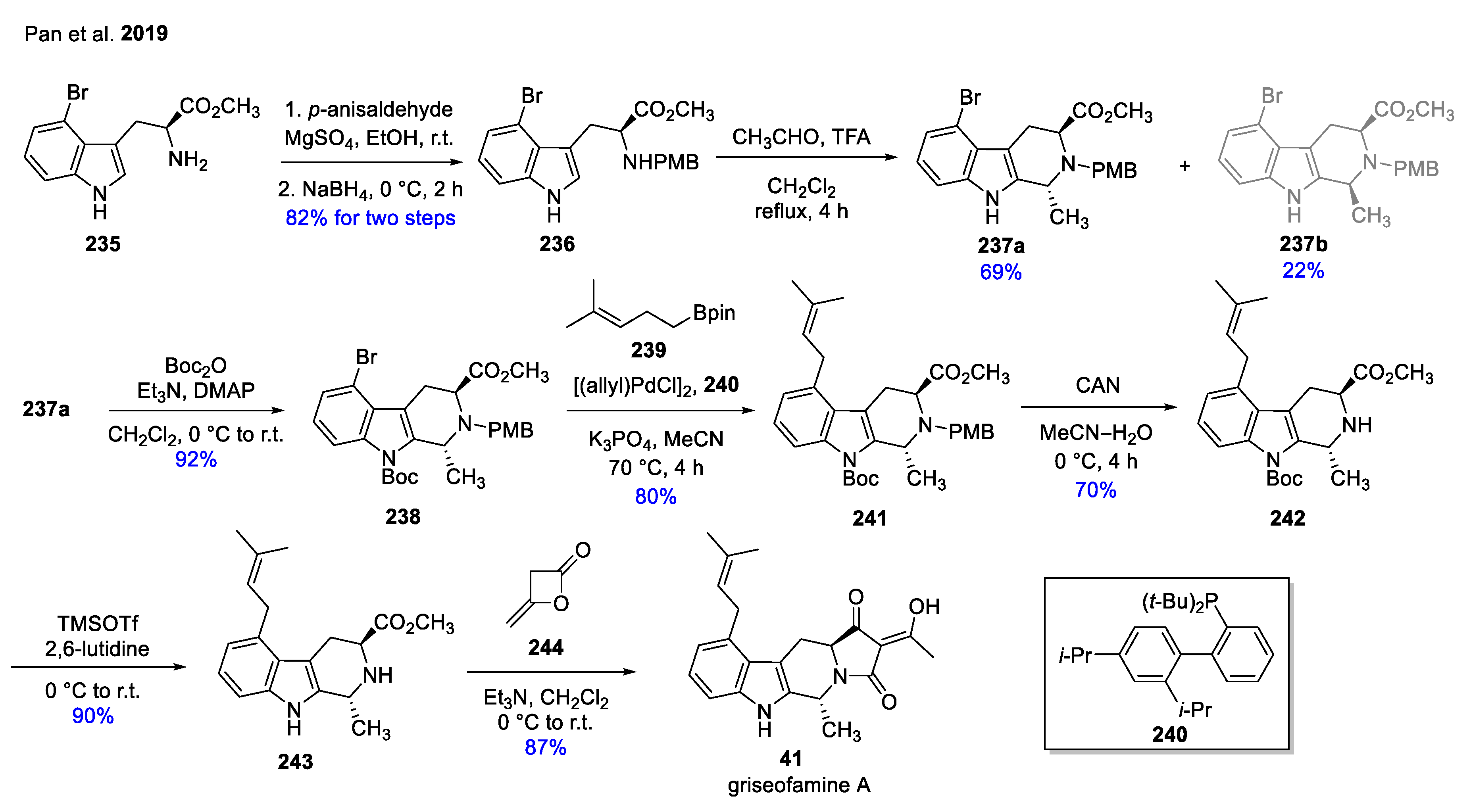{kind=link}
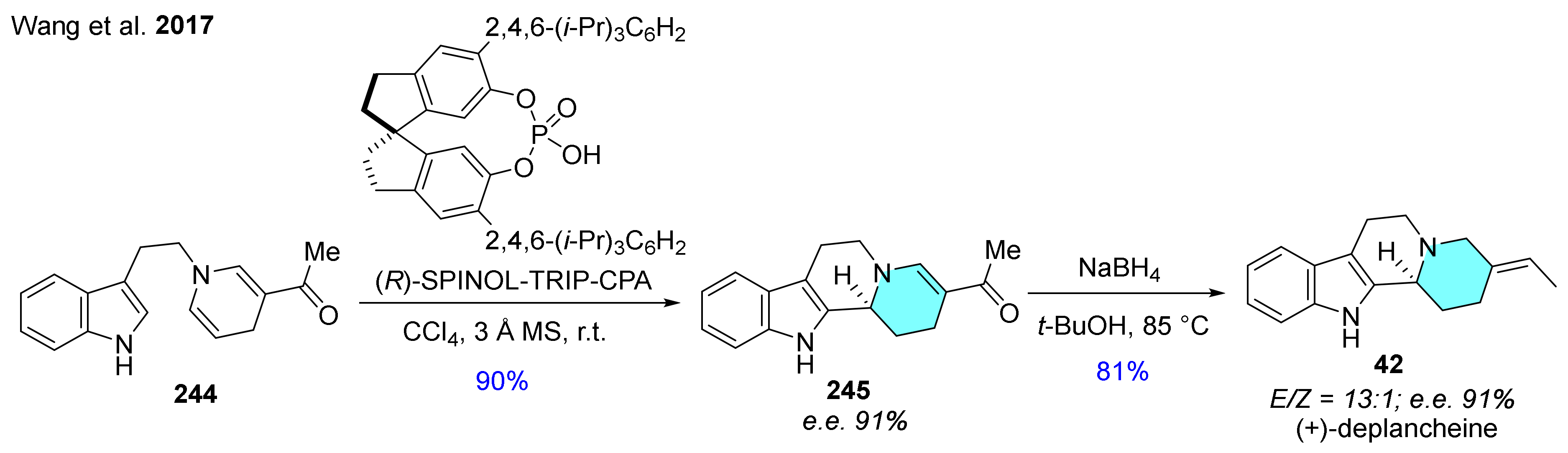{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
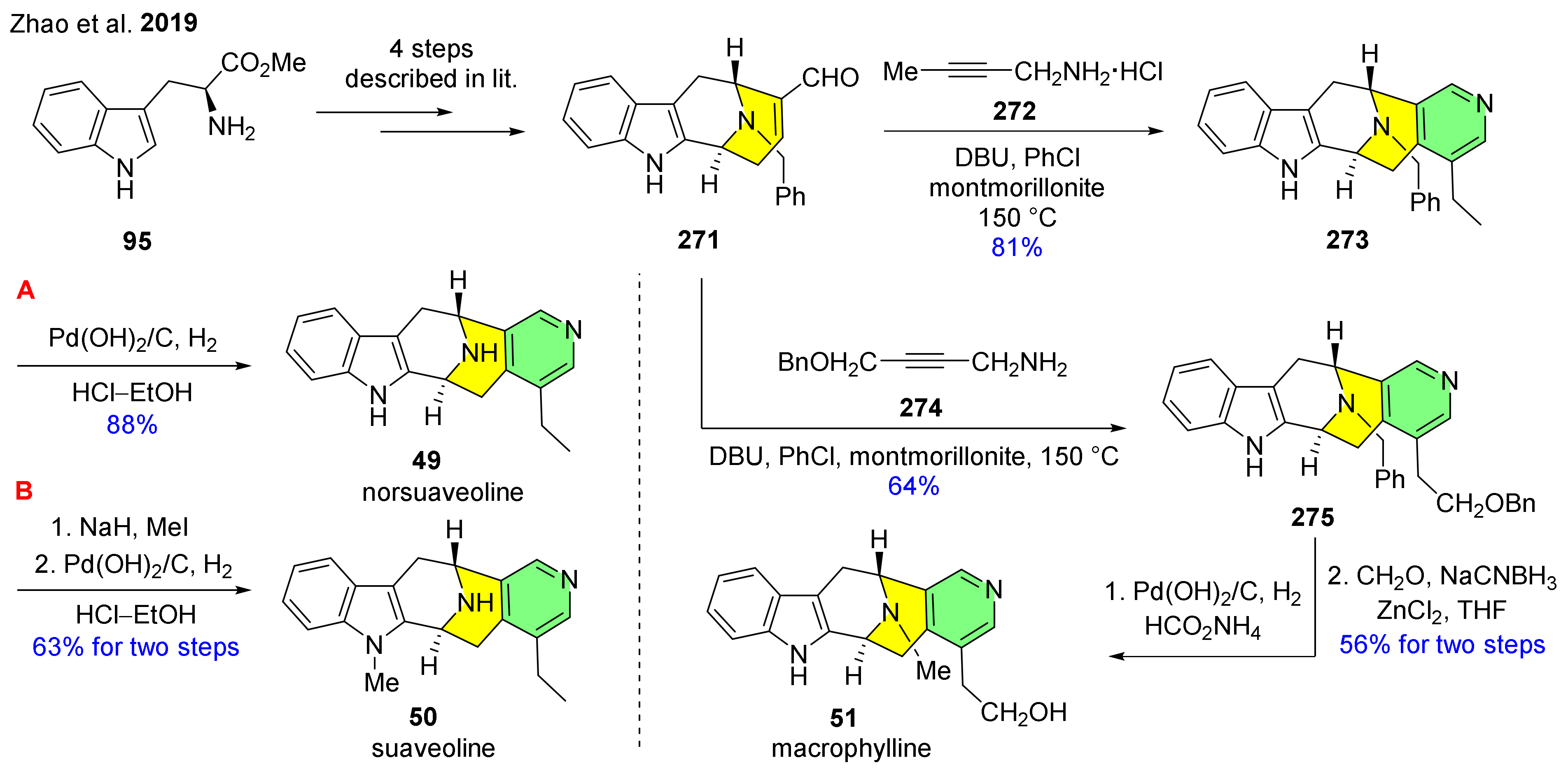{kind=link}
{kind=link}
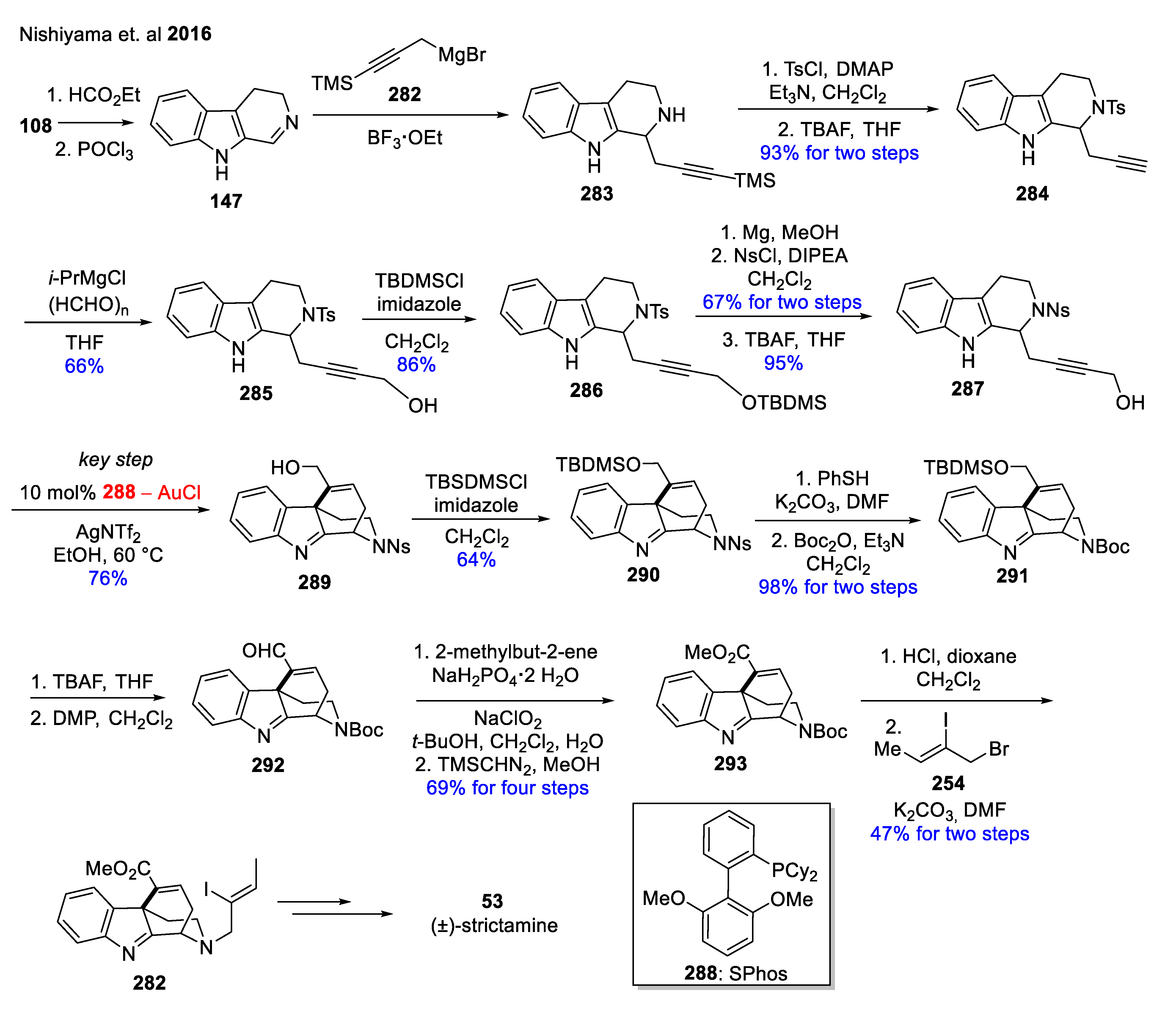{kind=link}
{kind=link}
{kind=link}
{kind=link}
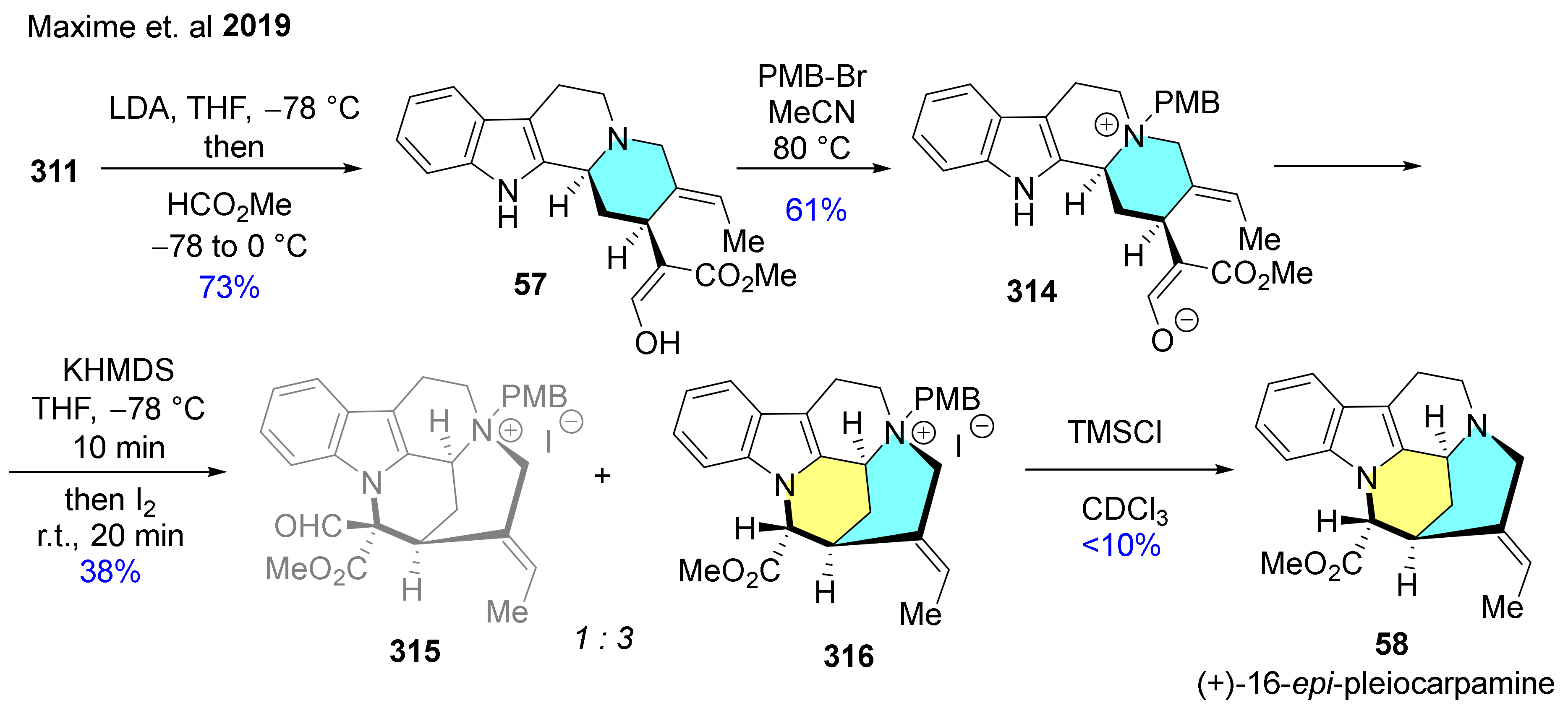{kind=link}
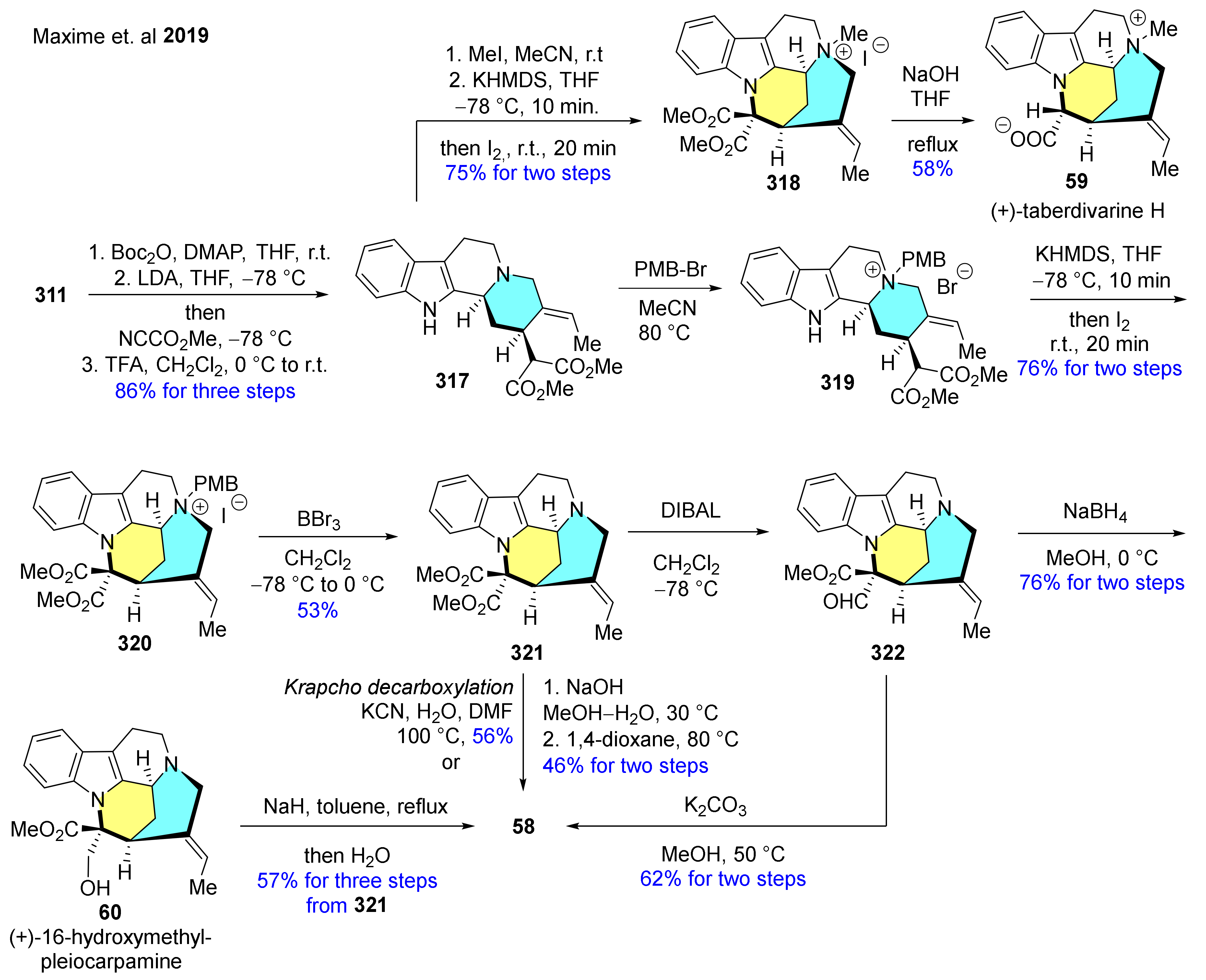{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
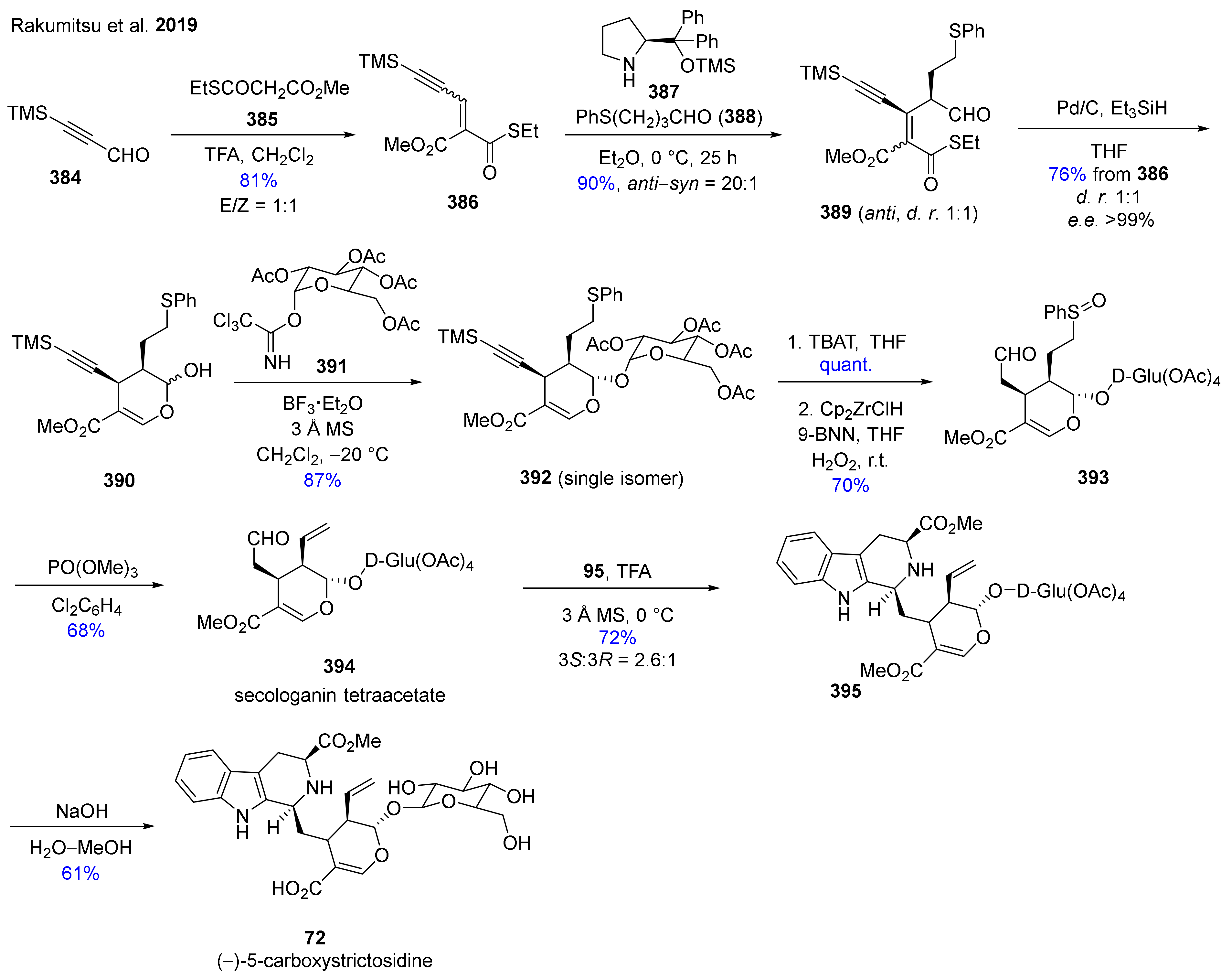{kind=link}
{kind=link}
Table 1.
Synthesized β-carbolines with their natural occurrence and bioactivity.
| ID | Name | Structure | Occurrence | Bioactivity |
|---|---|---|---|---|
| 1 | norharman(β-carboline) |  | Noctiluca milaris [10] Strychnos johnsonii [11] Lycoris longituba [12] Selaginella delicatula [13] Trichosanthes kirilowii Maxim [14] Rauwolfia caffra [15] Didemnum sp. [16] | MAO-B inhibitor11] |
| 2 3 4 5 6 | (−)-tetrahydroharman/ (S)-eleagnine (−)-komaroidine N-(+)-methyltetrahydro-harman (+)-1-ethyl-9-methyl-tetrahydro-β-carboline (+)-N-acetyl-komarodine |  | 2: Albizia polyphylla [17] 3, 6: N. komarovii and N.schoberi [18] 4: Petalostyles labicheoides [19] 5: analogues of natural products | not reported |
| 7 | stolonine C |  | Cnemidocarpa stolonifera [20] | apoptosis in PC3 human prostate cancer cell line [20] |
| 8 9 10 11 12 | eudistomin I eudistomin N eudistomin U eudistomin T isoeudistomin U |  | 8:Eudistoma olivaceum [21] 9: Eudistoma olivaceum [22] 10: Lissoclinum fragile [23] 11: Eudistoma olivaceum [24] 12: analogues of natural products | 8: antibacterial effect [25] 9: antiviral, antibacterial and antifungal activity [26] 11: antibiotic activity [27] |
| 13 14 15 16 | harmalan isoeudistomin M harman kumujian C |  | 13: soybean [28] Flindersia laevicarpa [29] 14: analogues of alcaloids 15: Flindersia Caevicarpa [29] 16: Picrasma quassioides [30] | 13: anti-neuroin-flammatory activities [28] 14: n.a. 15: vasorelaxant effect [31] binding affinity to imidazoline I2B receptors [32] 16: cytotoxic activity [33] AMP phosphodiesterase inhibitor [34] anti-inflammatory agent [35,36] |
| 17 18 19 20 21 | trigonostemine A trigonostemine B trigonostemine G pityriacitrin hyrtiosulawesine |  | 17, 18: Trigonostemone lii [37] 17, 18, 19: Trigonostemone filipes [38] 20: Malassezia furfur [39] Paracoccus marine bacteria [40] Dichotomomyces cejpii [41] 21: Hyrtio erectus [42] Alocasia macrorrhiza [43] | 17, 18, 19: cytotoxic activity [44] 20: antiproliferative activity [45] anticancer activity [46,47] 21: antiproliferative activity [48,49] antimalarial activity [46] antiphospholipase A2 [50] and antioxidant activity [46] |
| 22 23 | alangiobussine alangiobussinine |  | Alangium bussyanum [51] | not reported |
| 24 | haploscleridamine |  | Sponge of the order Haplosclerida [52] | inhibitor of cathepsin K [52] |
| 25 | (±)-peharmaline A |  | Peganum harmala [53] | cytotoxic activity (HL-60, PC-3, SGC-7901) [54] |
| 26 | komavine |  | Nitraria komarorii [55] Nitraria schoberi [55] | not reported |
| 27 28 29 30 | marinacarboline A marinacarboline B marinacarboline C marinacarboline D |  | Marinactinospora thermotolerans SCSIO 00652 [56] | antiplasmodial activity [56] cytotoxic activity [57] |
| 31 | metatacarboline A |  | Mycena metata [58] | not reported |
| 32 33 34 35 | metatacarboline C metatacarboline D metatacarboline E metatacarboline F |  | Mycena metata [58] | 32, 34: antiproliferative activity, proapoptotic effect [59] |
| 36 | 6-hxdroxy- metatacarboline D |  | Mycena metata [58] | not reported |
| 37 | shishijimicin A |  | Didemnum proliferum [60] | antitumor activity (P388 leukemia cells) [60] |
| 38 | canthine |  | basic framework of canthin-6-ones, which are widely distributed in nature [61] | not reported |
| 39 | harmicine |  | Kopsia griffithii [62] | not reported |
| 40 | cordatanine |  | Drymaria cordata [63] Drymaria diandra [64,65] | anti-HIV activity [64] |
| 41 | griseofamine A |  | Penicillium griseofulvum [66] | antibacterial activity [67] |
| 42 | (+)-deplancheine |  | Alstonia deplanchei [68] | not reported |
| 43 | (−)-geissoschizol |  | Tabernaemontana bufalina [69] Hunteria zeylanica [70] | not reported |
| 44 | chaetogline E |  | modified Epinephelus drummondhayi with a silent fungal gene from Chaetomium globosum [71] | not reported |
| 45 | chaetogline F |  | modified Epinephelus drummondhayi with a silent fungal gene from Chaetomium globosum [71] | antibacterial activity [71] |
| 46 | 6-oxofascaplysin |  | Hyrtios sp. [72] | weak cytotoxic activity [73] |
| 47 | fascaplysin |  | Fascaplisynopsis sp [74] | anticancer activity [73,75] |
| 48 | evodiamine |  | Evodia rutaecarpa [76] | anticancer anti-inflammatory antimicrobial and many others [77] |
| 49 50 51 | norsuaveoline suaveoline marcophylline |  | 49:Rauvolfia caffra [78] 50: Rauwolfia suaveolens S. Moore (apocynaceae) [79] 51: Rauwolfia macrophylla [80] | 49: not reported 50: acetylcholinesterase inhibitor [81] 51: not reported |
| 52 | (S)-(−)-decarbo-methoxy-dihydro-gambirtannine |  | enantiomer of 52 was isolated from Hunteria zeylanica [82] | not reported |
| 53 | (+)-strictamine |  | Alstonis scholaris Rhazya strictu [83,84] | antitumor, antiviral anti-inflammatory antibiosis activities [83,84,85] |
| 54 | (±)-arbornamine |  | Kopsia arborea [86] | not reported |
| 55 | (+)-tacamonine |  | Tabernaemontana eglandulosa [87] | not reported |
| 56 | (−)-17-nor-excelisidine |  | Alstonia scholaris [85] | antiviral activity [85] |
| 57 | (+)-geissoschizine |  | Rhazya stricta [88] | not reported |
| 58 59 60 61 62 63 | (+)-16-epi-pleiocarpamine (+)-taberdivarine H (+)-16-hydroxymethyl-pleiocarpamine (+)-pleiocarpamine (±)-normavacurine (±)-C-mavacurine |  | 58: Ervatamia cumingiana [89] Pleiocarpa pycnantha [90] 59: Tabernaemontana officinalis [91] 60: Kopsia singapurensis [92] Kopsia teoi [93] 62: Alstonia scholaris [94] 63: Strychnos guianensis [95] | 61, 58: anticholinergic activity [96] 59: cytotoxic activity [91] 60: not reported 62: antibacterial activity [94] 63: not reported |
| 64 65 66 67 | (+)-19-oxoeburnamine (+)-19-OH-eburnamine 19-(S)-hydroxy-Δ14-vincamone (–)-19-hydroxy-Δ14-eburnamonine |  | Kopsia jasminiflora [97] Kopsia pauciflora [98] | not reported closely related analogues show antitumor activity |
| 68 | (+)-vallesamidine |  | Vallesia dichotoma [99] | not reported |
| 69 | (+)-14,15-dehydro-strempeliopine |  | Schizozygia caffaeoides [100] | not reported |
| 70 | (+)-peganumine A |  | Peganum harmala L. [101] | cytotoxic activity (MCF-70, PC-3, HepG2, HL-60) [101] |
| 71 | reserpine |  | Rauwolfia serpentina [102] | Commercially available drug for the treatment of high blood pressure [103] |
| 72 | (–)-5-carboxystrictosidine |  | Rhazya orientalis [104] Guenttarda platypoda [105] Uncaria tomentosa [106] Ophiorrhiza nutans [107] | not reported |
| 73 | (–)-rubenine |  | Adina rubescens [108] | not reported |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Szabó, T.; Volk, B.; Milen, M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. https://doi.org/10.3390/molecules26030663
AMA Style
Szabó T, Volk B, Milen M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules. 2021; 26(3):663. https://doi.org/10.3390/molecules26030663
Chicago/Turabian StyleSzabó, Tímea, Balázs Volk, and Mátyás Milen. 2021. "Recent Advances in the Synthesis of β-Carboline Alkaloids" Molecules 26, no. 3: 663. https://doi.org/10.3390/molecules26030663