
Comparative Analysis of the Complete Mitochondrial Genomes of Apium graveolens and Apium leptophyllum Provide Insights into Evolution and Phylogeny Relationships
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Analysis of Mitochondrial Genome Assembly and Annotation Results
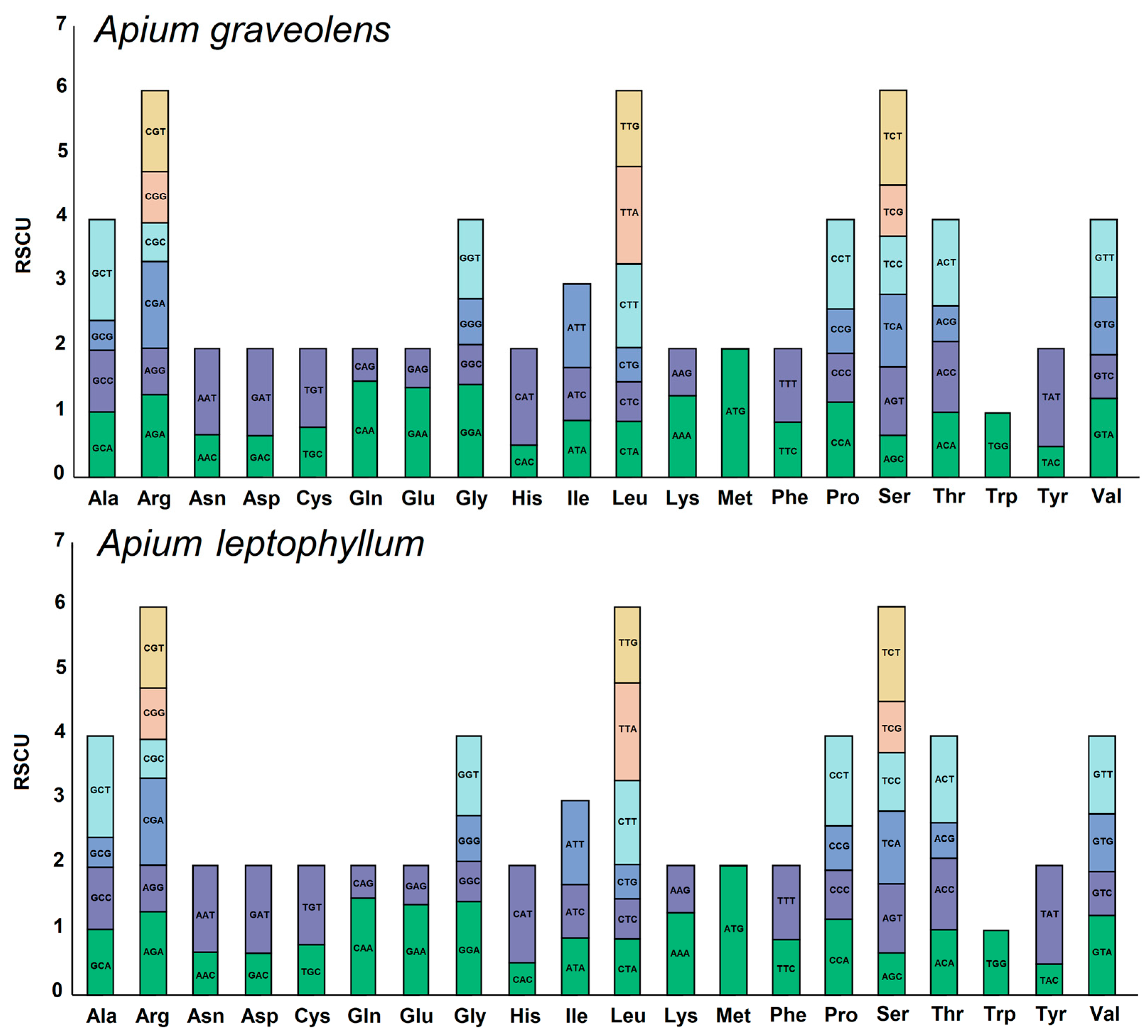
2.2. RSCU Analysis
2.3. SSRs and Repeat Sequence Analysis
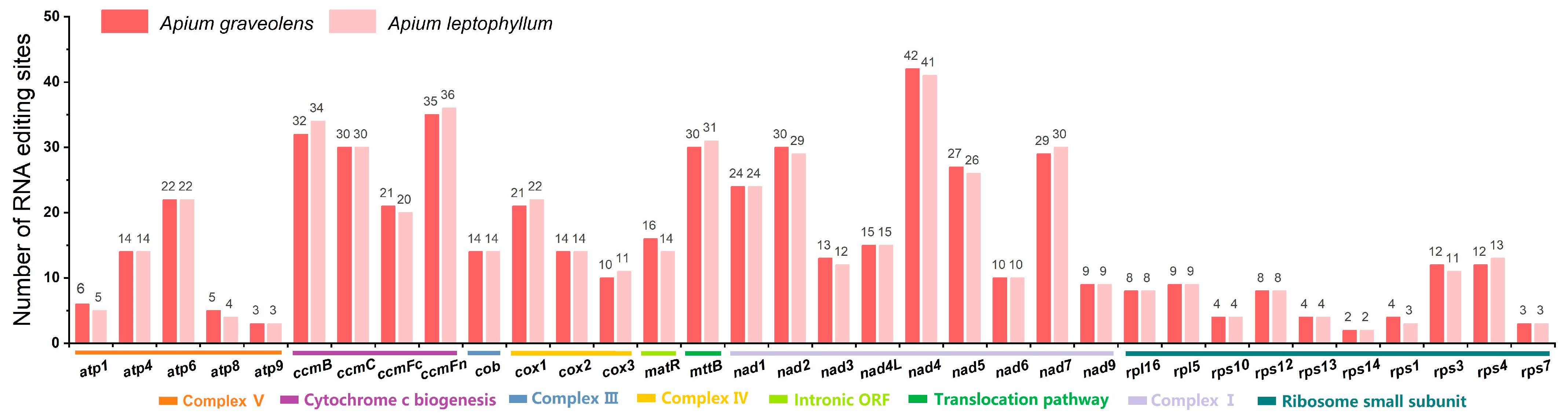
2.4. The Prediction of RNA Editing
2.5. Structural Comparison of A. graveolens and A. leptophyllum
2.6. Sequence Similarity between Mitochondrial and Chloroplast Genomes
2.7. The Ka/Ks Ratio and Nucleotide Diversity (Pi) Values
2.8. Comparative Genomics Analysis
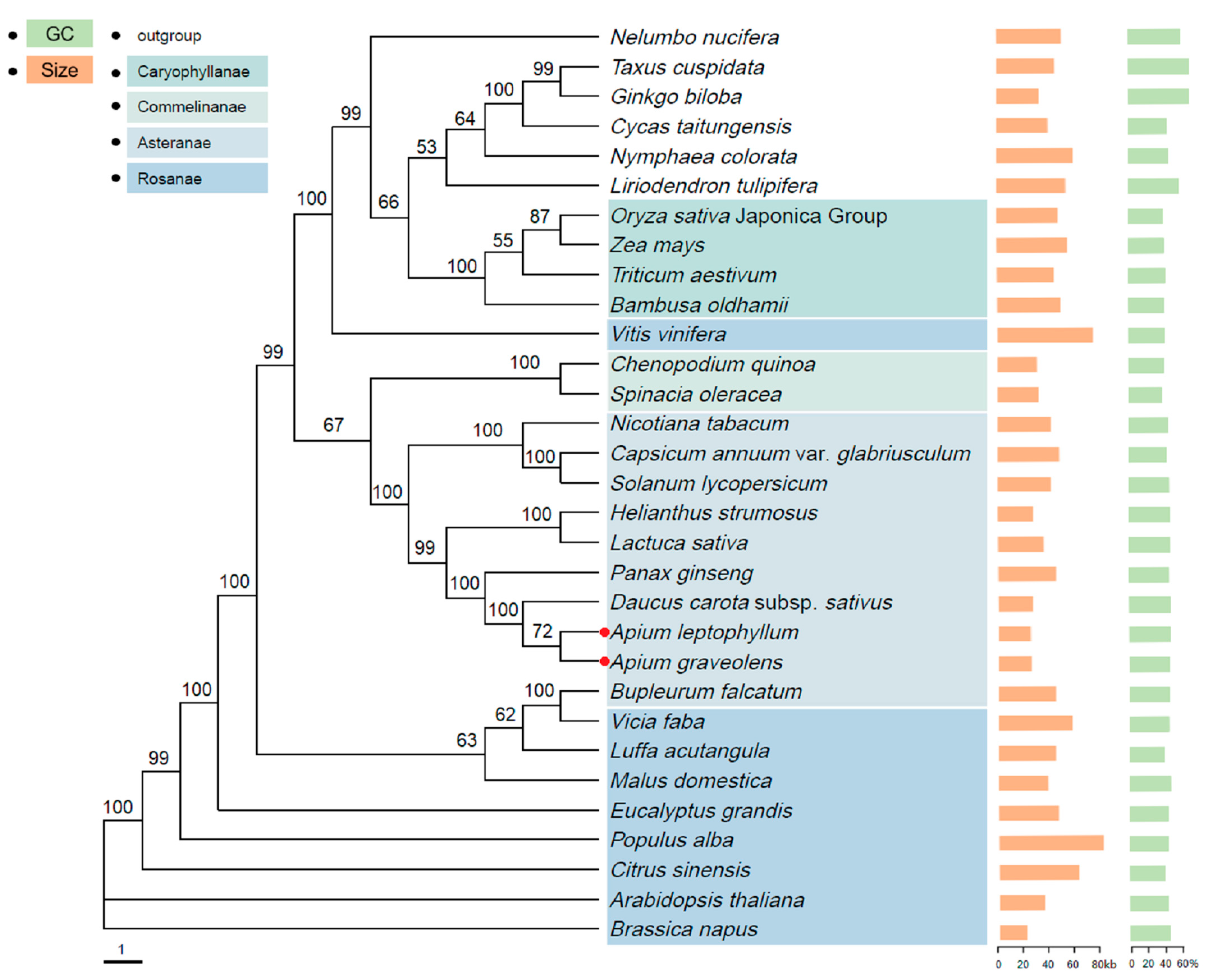
2.9. Phylogenetic Relationship
3. Discussion
4. Materials and Methods
4.1. Materials, DNA Extraction, and Sequencing
4.2. Mitochondrial Genome Assembly
4.3. Genome Annotations and Analysis
4.4. RNA Editing Analysis and Characteristic Analysis of Mitochondrial Genome
4.5. Comparative Analysis of Mitochondrial Genome
4.6. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes though this be madness, yet there’s method in’t. J. Exp. Bot. 2010, 61, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Liao, X.Z.; Zhang, X.N.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Niu, L.; Zhang, Y.; Yang, C.; Yang, J.; Ren, W.; Zhong, X.; Zhao, Q.; Xing, G.; Zhao, Y.; Yang, X. Complete mitochondrial genome sequence and comparative analysis of the cultivated yellow nutsedge. Plant Genome 2022, 15, e20239. [Google Scholar] [CrossRef] [PubMed]
- Daloso, D.M.; Müller, K.; Obata, T.; Florian, A.; Tohge, T.; Bottcher, A.; Riondet, C.; Bariat, L.; Carrari, F.; Nunes-Nesi, A.; et al. Thioredoxin, a master regulator of the tricarboxylic acid cycle in plant mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, E1392–E1400. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Dong, S.; Chen, L.; Liu, Y.; Wang, Y.; Zhang, S.; Yang, L.; Lang, X.; Zhang, S. The draft mitochondrial genome of Magnolia biondii and mitochondrial phylogenomics of angiosperms. PLoS ONE 2020, 15, e0231020. [Google Scholar] [CrossRef]
- Smith, D.R.; Keeling, P.J. Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. USA 2015, 112, 10177–10184. [Google Scholar] [CrossRef]
- Noutahi, E.; Calderon, V.; Blanchette, M.; El-Mabrouk, N.; Lang, B.F. Rapid genetic code evolution in green algal mitochondrial genomes. Mol. Biol. Evol. 2019, 36, 766–783. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kang, Y.; Kim, M.K.; Lee, J.; Park, H.; Kim, J.H.; Lee, H. The complete mitochondrial genome of an Antarctic moss Syntrichia filaris (Müll.Hal.) R.H. Zander. Mitochondrial DNA B Resour. 2016, 27, 2779–2780. [Google Scholar] [CrossRef]
- Yurina, N.P.; Odintsova, M.S. Mitochondrial genome structure of photosynthetic eukaryotes. Biochemistry 2016, 81, 101–113. [Google Scholar] [CrossRef]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Moreno, L.; González, V.M.; Benjak, A.; Martí, M.C.; Puigdomènech, P.; Aranda, M.A.; Garcia-Mas, J. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genome 2011, 12, 424. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Lauren, C.; Warren, R.L.; Heather, K.; Eva, T.; MacLeod, T.; Pleasance, S.; Pandoh, P.; Zhao, Y.; Coope, R.J.; et al. Complete mitochondrial genome of a gymnosperm, Sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 2020, 12, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Goremykin, V.V.; Lockhart, P.J.; Viola, R.; Velasco, R. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 2012, 71, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- Zou, Y.; Zhu, W.D.; Sloan, D.B.; Wu, Z.Q. Long-read sequencing characterizes mitochondrial and plastid genome variants in Arabidopsis msh1 mutants. Plant J. 2022, 112, 738–755. [Google Scholar] [CrossRef]
- Van de Paer, C.; Bouchez, O.; Besnard, G. Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 2018, 18, 407–423. [Google Scholar] [CrossRef]
- Preston, M.D.; Campino, S.; Assefa, S.A.; Echeverry, D.F.; Ocholla, H.; Amambua-Ngwa, A.; Stewart, L.B.; Conway, D.J.; Borrmann, S.; Michon, P.; et al. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat. Commun. 2014, 5, 4052. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, X.; Li, Z.; Song, Y.; Sun, Z. Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef]
- Varré, J.S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Yu, W.; Ge, X.; Li, H.; Sun, C.; Mao, X.; Yuan, K. Root exudation of prometryn and its metabolites from Chinese celery (Apium graveolens). J. Pestic. Sci. 2022, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, X.; Zhou, J.; Sun, Y.; Du, J.; Wang, Z.; Luo, Y.; Zhang, Y.; Chen, Q.; Wang, Y.; et al. Genome-wide identification and analysis of terpene synthase (TPS) genes in celery reveals their regulatory roles in terpenoid biosynthesis. Front. Plant Sci. 2022, 13, 1010780. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Hou, X.L.; Wang, F.; Tan, G.F.; Xu, Z.S.; Xiong, A.S. Advances in the research of celery, an important Apiaceae vegetable crop. Crit. Rev. Biotechnol. 2018, 38, 172–183. [Google Scholar] [CrossRef]
- Asamenew, G.; Tadesse, S.; Asres, K.; Mazumder, A.; Bucar, F. A study on the composition, antimicrobial and antioxidant activities of the leaf essential oil of Apiumleptophyllum (Pers.) Benth. Growing in Ethiopia. Ethiop. Pharm. J. 2008, 26, 94–99. [Google Scholar]
- Li, J.; Tang, H.; Luo, H.; Tang, J.; Zhong, N.; Xiao, L. Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 2023, 14, 1117002. [Google Scholar] [CrossRef]
- Chang, S.; Yang, T.; Du, T.; Huang, Y.; Chen, J.; Yan, J.; He, J.; Guan, R. Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in brassica. BMC Genom. 2011, 12, 497. [Google Scholar] [CrossRef]
- Myszczyński, K.; Ślipiko, M.; Sawicki, J. Potential of transcript editing across mitogenomes of early land plants Shows Novel and Familiar Trends. Int. J. Mol. Sci. 2019, 20, 2963. [Google Scholar] [CrossRef]
- Liu, Y.; Cox, C.J.; Wang, W.; Goffinet, B. Mitochondrial phylogenomics of early land plants: Mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 2014, 63, 862–878. [Google Scholar] [CrossRef]
- Verhage, L. Targeted editing of the Arabidopsis mitochondrial genome. Plant J. 2020, 104, 1457–1458. [Google Scholar] [CrossRef]
- Fang, B.; Li, J.; Zhao, Q.; Liang, Y.; Yu, J. Assembly of the complete mitochondrial genome of chinese plum (Prunus salicina): Characterization of genome recombination and RNA editing sites. Genes 2021, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Xiao, G.; Liu, H.; Zhang, L.; Zhao, L.; Tang, M.; Huang, S.; An, Y.; Yu, J. Two pivotal RNA editing sites in the mitochondrial atp1 mRNA are required for ATP synthase to produce sufficient ATP for cotton fiber cell elongation. New Phytol. 2018, 218, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Giegé, P.; Brennicke, A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 1999, 96, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Naka-zono, M.; Hirai, A. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evo-lution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Hu, J.H.; Gui, S.T.; Zhu, Z.X.; Wang, X.; Ke, W.; Ding, Y. Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred Lotus (Nelumbo nucifera). PLoS ONE 2015, 10, e0143765. [Google Scholar] [CrossRef]
- Ammiraju, J.S.S.; Zuccolo, A.; Yu, Y.; Song, X.; Piegu, B.; Chevalier, F.; Walling, J.G.; Ma, J.; Talag, J.; Brar, D.S.; et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007, 52, 34. [Google Scholar] [CrossRef]
- Han, F.; Qu, Y.; Chen, Y.; Xu, L.; Bi, C. Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 2022, 13, 1031769. [Google Scholar] [CrossRef]
- Bi, C.; Qu, Y.; Hou, J.; Wu, K.; Ye, N.; Yin, T. Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 2022, 13, 914635. [Google Scholar] [CrossRef]
- Timmis, J.N.; Aylife, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitogenomes of Suaeda Glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. Biomed. Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, H.S.; An, H.M. Chloroplast DNA-based genetic variation of Rosa roxburghii in Southwest China: Phylogeography and conservation implications. Hortic. Plant J. 2021, 7, 286–294. [Google Scholar] [CrossRef]
- Xiong, Y.; Yu, Q.; Xiong, Y.; Zhao, J.; Lei, X.; Liu, L.; Liu, W.; Peng, Y.; Zhang, J.; Li, D. The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 2022, 12, 802321. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar]
- Li, M.Y.; Zhang, R.; Li, R.; Zheng, K.M.; Xiao, J.C.; Zheng, Y.X. Analyses of chloroplast genome of Eutrema japonicum provide new Insights into the evolution of Eutrema Species. Agronomy 2021, 11, 2546. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, M.; Li, W.; Zhou, J.; Han, Q.; Lu, W.; Luo, Q.; Zhu, S.; Xiong, A.; Tan, G.; et al. Comparative Analysis of the Complete Mitochondrial Genomes of Apium graveolens and Apium leptophyllum Provide Insights into Evolution and Phylogeny Relationships. Int. J. Mol. Sci. 2023, 24, 14615. https://doi.org/10.3390/ijms241914615
Li X, Li M, Li W, Zhou J, Han Q, Lu W, Luo Q, Zhu S, Xiong A, Tan G, et al. Comparative Analysis of the Complete Mitochondrial Genomes of Apium graveolens and Apium leptophyllum Provide Insights into Evolution and Phylogeny Relationships. International Journal of Molecular Sciences. 2023; 24(19):14615. https://doi.org/10.3390/ijms241914615
Chicago/Turabian StyleLi, Xiaoyan, Mengyao Li, Weilong Li, Jin Zhou, Qiuju Han, Wei Lu, Qin Luo, Shunhua Zhu, Aisheng Xiong, Guofei Tan, and et al. 2023. "Comparative Analysis of the Complete Mitochondrial Genomes of Apium graveolens and Apium leptophyllum Provide Insights into Evolution and Phylogeny Relationships" International Journal of Molecular Sciences 24, no. 19: 14615. https://doi.org/10.3390/ijms241914615