
Genome Assembly of Cordia subcordata, a Coastal Protection Species in Tropical Coral Islands
Abstract
:1. Introduction
2. Results
2.1. Genome Sequencing and Assembly
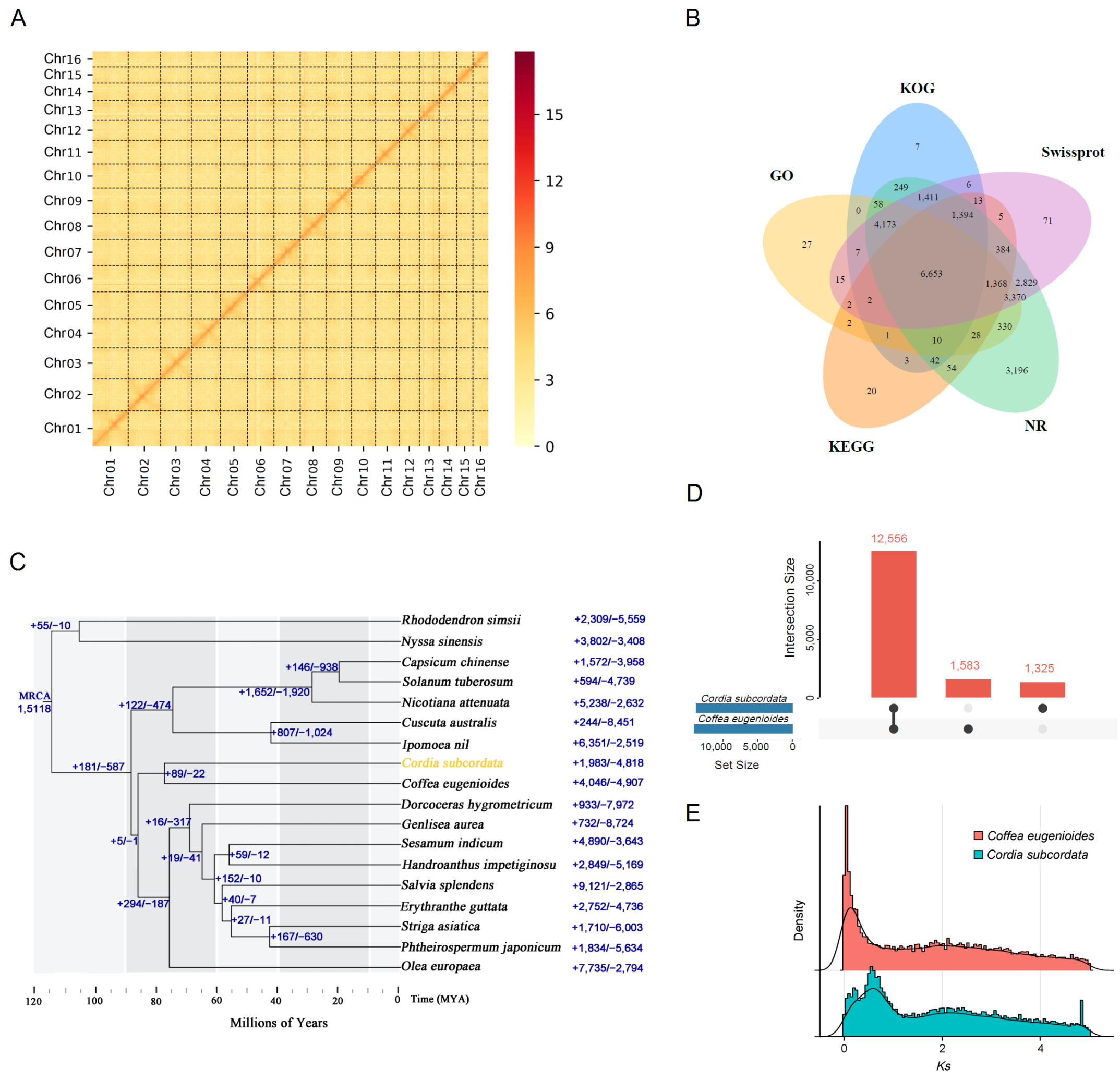
2.2. Completeness of the Genome and Quality Evaluation
2.3. Repeat Sequence Prediction
2.4. Gene Prediction and Annotation
2.5. Orthologous Gene Identification and Phylogenetic Analysis
2.6. Whole Genome Duplication and Gene Duplication
3. Discussion
4. Materials and Methods
4.1. Sequencing
4.2. Assembly
4.3. Annotation
4.4. Orthologous Gene Group Identification and Whole Genome Duplication Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xing, F.W.; Deng, S.W. Flora of the South China Sea Island; China Forestry Press: Beijing, China, 2018. [Google Scholar]
- Liu, J.X.; Xi, Y.Z.; Ning, J.C.; Zhang, J.M.; Li, Y.X.; Zhao, Y.Y.; Sun, X.H. Pollen morphology and exine ultrastructure of genus Cordia in Boraginaceae in China. Acta. Bot. Sin. 2001, 43, 893–898. [Google Scholar]
- Bouby, L.; Bouchette, A.; Figueiral, I. Sebesten fruits (Cordia myxa L.) in Gallia Narbonensis (Southern France): A trade item from the Eastern Mediterranean? Veget. Hist. Archaeobot. 2011, 20, 397–404. [Google Scholar] [CrossRef]
- Wang, W. An enumeration of the Boraginaceous plants collected by H.smith from China during 1921–22, 1924 and 1934. Bull. Bot. Res. 1993, 13, 1–10. [Google Scholar]
- Matias, E.F.F.; Alves, E.F.; Silva, M.K.D.N.; Carvalho, V.R.D.A.; Coutinho, H.D.M.; Costa, J.G.M. The genus Cordia: Botanists, ethno, chemical and pharmacological aspects. Rev. Bras. Farmacogn. 2015, 25, 542–552. [Google Scholar] [CrossRef]
- Huang, Z.G.; Zhang, W.Q. A discussion on the quaternary climate record from the coral reef in tropical China. Trop. Geogr. 2008, 2, 11–15. [Google Scholar]
- Jian, S.G. Vegetation of tropical coral islands in China. Guihaia 2020, 40, 443. [Google Scholar]
- Wu, S.H.; Chen, H.W.; Jian, S.G.; Liu, H.; Zhang, W.; Ren, H. The biological characteristics of Cordia subcordata on tropical coral island in China. Ecol. Sci. 2017, 36, 57–63. [Google Scholar]
- Oza, M.J.; Kulkarni, Y.A. Traditional uses, phytochemistry and pharmacology of the medicinal species of the genus Cordia (Boraginaceae). J. Pharm. Pharmacol. 2017, 69, 755–789. [Google Scholar] [CrossRef]
- Nisha, M.; Chandru, N.; Pradeep, P.; Selvarasu, P.; Surendra Kumar, M.; Latha, S.T.; Astalakshmi, N. A review on phytochemical and pharmacological activity of Cordia sebestena. Int. J. Pharm. Sci. Rev. Res. 2022, 73, 156–161. [Google Scholar]
- Liu, D.M.; Chen, H.F.; Wang, F.G.; Yi, Q.F.; Xing, F.W. Investigation of introduced plants in Nansha islands and reefs, China. J. Trop. Subtrop. Bot. 2015, 23, 167–175. [Google Scholar]
- Wang, X.P.; Wen, M.H.; Wu, M.S.; Zhang, D.X. Cordia subcordata (Boraginaceae), a distylous species on oceanic coral islands, is self-compatible and pollinated by a passerine bird. Plant Ecol. Evol. 2020, 153, 361–372. [Google Scholar] [CrossRef]
- Xiong, Y.P.; Chen, X.H.; Wu, K.L.; da Silva, J.A.T.; Zeng, S.J.; Ma, G.H. Shoot organogenesis and plant regeneration in Cordia subcordata Lam. In Vitro Cell. Dev. Biol. Plant 2022, 58, 392–398. [Google Scholar] [CrossRef]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.K.; Liu, M.; Chen, Y.W.; Gui, J.S.; Fang, D.M.; Chen, X.L.; Yang, T.; He, C.Z.; Cheng, L.; Yang, J.L.; et al. Chromosome-scale genomes of commercial timber trees (Ochroma pyramidale, Mesua ferrea, and Tectona grandis). Sci. Data 2023, 10, 512. [Google Scholar] [CrossRef]
- Wang, Z.F.; Zhang, X.; Lei, W.X.; Zhu, H.; Wu, S.D.; Liu, B.B.; Ru, D.F. Chromosome-level genome assembly and population genomics of Robinia pseudoacacia reveal the genetic basis for its wide cultivation. Commun. Biol. 2023, 6, 797. [Google Scholar] [CrossRef]
- Qin, Y.; Zhao, H.; Han, H.W.; Zhu, G.P.; Wang, Z.S.; Li, F.D. Chromosome-level genome assembly and population genomic analyses reveal geographic variation and population genetic structure of Prunus tenella. Int. J. Mol. Sci. 2023, 24, 11735. [Google Scholar] [CrossRef]
- Cohen, J.I. A phylogenetic analysis of morphological and molecular characters of Boraginaceae: Evolutionary relationships, taxonomy, and patterns of character evolution. Cladistics 2014, 30, 139–169. [Google Scholar] [CrossRef]
- Hasenstab-Lehman, K.E. Phylogenetics of the Borage family: Delimiting Boraginales and assessing closest relatives. Aliso A J. Syst. Florist. Bot. 2017, 35, 41–49. [Google Scholar] [CrossRef]
- Alawfi, M.S.; Albokhari, E.J. Comparative chloroplast genomics reveals a unique gene inversion in two Cordia trees (Cordiaceae). Forests 2023, 14, 1778. [Google Scholar] [CrossRef]
- Alshegaihi, R.M.; Mansour, H.; Alrobaish, S.A.; Al Shaye, N.A.; Abd El-Moneim, D. The first complete chloroplast genome of Cordia monoica: Structure and comparative Analysis. Genes 2023, 14, 976. [Google Scholar] [CrossRef]
- Sun, X.; Wang, G.; Yang, J.; Yu, W.; Xu, J.; Tang, B.; Ding, G.; Zhang, D. Whole genome evaluation analysis and preliminary assembly of Oratosquilla oratoria (Stomatopoda: Squillidae). Mol. Biol. Rep. 2023, 50, 4165–4173. [Google Scholar] [CrossRef]
- Tang, C.Y.; Li, S.; Wang, Y.T.; Wang, X. Comparative genome/transcriptome analysis probes Boraginales’ phylogenetic position, WGDs in Boraginales, and key enzyme genes in the alkannin/shikonin core pathway. Mol. Ecol. Resour. 2020, 20, 228–241. [Google Scholar] [CrossRef]
- Auber, R.P.; Suttiyut, T.; McCoy, R.M.; Ghaste, M.; Crook, J.W.; Pendleton, A.L.; Widhalm, J.R.; Wisecaver, J.H. Hybrid de novo genome assembly of red gromwell (Lithospermum erythrorhizon) reveals evolutionary insight into shikonin biosynthesis. Hortic. Res. 2020, 7, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Kobrlova, L.; Hrones, M. First insights into the evolution of genome size in the borage family: A complete data set for Boraginaceae from the Czech Republic Bot. J. Linn. Soc. 2019, 189, 115–131. [Google Scholar] [CrossRef]
- Fang, Y.; Jiang, J.; Hou, X.; Guo, J.; Li, X.; Zhao, D.; Xie, X. Plant protein-coding gene families: Their origin and evolution. Front. Plant Sci. 2022, 13, 995746. [Google Scholar] [CrossRef]
- Tao, W.; Li, R.; Li, T.; Li, Z.; Li, Y.; Cui, L. The evolutionary patterns, expression profiles, and genetic diversity of expanded genes in barley. Front. Plant Sci. 2023, 14, 1168124. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.H.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kokabu, Y.; Chaki, K.; Yoshimoto, T.; Ohgaki, M.; Yoshida, S.; Kato, N.; Koyama, T.; Sato, F. Isoquinoline alkaloid biosynthesis is regulated by a unique bHLH-Type transcription factor in Coptis japonica. Plant Cell Physiol. 2011, 52, 1131–1141. [Google Scholar] [CrossRef]
- Singh, S.; Pathak, N.; Fatima, E.; Negi, A.S. Plant isoquinoline alkaloids: Advances in the chemistry and biology of berberine. Eur. J. Med. Chem. 2021, 226, 113839. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.Y.; Sun, Z.D.; Lai, Z.G.; Yang, S.Z.; Chen, H.Z.; Yang, X.H.; Tao, J.R.; Tang, X.M. Determination of the miRNAs related to bean pyralid larvae resistance in soybean using small RNA and transcriptome sequencing. Int. J. Mol. Sci. 2019, 20, 2966. [Google Scholar] [CrossRef]
- Ren, Z.Y.; Fang, M.K.; Muhae-Ud-Din, G.; Gao, H.F.; Yang, Y.Z.; Liu, T.G.; Chen, W.Q.; Gao, L. Metabolomics analysis of grains of wheat infected and noninfected with Tilletia controversa Kuhn. Sci. Rep. 2021, 11, 18876. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Wu, P.F.; Yao, X.H.; Sheng, Y.; Zhang, C.C.; Lin, P.; Wang, K.L. Integrated transcriptome and metabolome analysis reveals key metabolites involved in Camellia oleifera defense against anthracnose. Int. J. Mol. Sci. 2022, 23, 536. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2016, 220, 655–658. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Jin, J.J.; Sarojam, R.; Ramachandran, S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 2019, 11, 2078–2098. [Google Scholar] [CrossRef] [PubMed]
- Salazar, D.; Lokvam, J.; Mesones, I.; Pilco, M.V.; Zuñiga, J.M.A.; de Valpine, P.; Fine, P.V.A. Origin and maintenance of chemical diversity in a species-rich tropical tree lineage. Nat. Ecol. Evol. 2018, 2, 983–990. [Google Scholar] [CrossRef]
- Hallam, A.; Read, J. Do tropical species invest more in anti-herbivore defence than temperate species? A test in Eucryphia (Cunoniaceae) in eastern Australia. J. Trop. Ecol. 2006, 22, 41–51. [Google Scholar] [CrossRef]
- Abdel-Aleem, E.R.; Attia, E.Z.; Farag, F.F.; Samy, M.N.; Desoukey, S.Y. Total phenolic and flavonoid contents and antioxidant, anti-inflammatory, analgesic, antipyretic and antidiabetic activities of Cordia myxa L. leaves. Clin. Phytosci. 2019, 5, 29. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Lu, C.Q.; Wang, Y.; Wang, Q. Phenolpropane compounds of Cordia dichotoma and their anticomplementary activities. Chem. Nat. Compd. 2021, 57, 169–170. [Google Scholar] [CrossRef]
- Raghuvanshi, D.; Sharma, K.; Verma, R.; Kumar, D.; Kumar, H.; Khan, A.; Valko, M.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; et al. Phytochemistry, and pharmacological efficacy of Cordia dichotoma G. Forst. (Lashuda): A therapeutic medicinal plant of Himachal Pradesh. Biomed. Pharmacother. 2022, 153, 113400. [Google Scholar] [CrossRef]
- Ahmed Bokhari, S.W.; Sharif, H.; Umer Gilani, S.M.; Ali, S.T.; Ahmed, S.; Ahmed Siddiqui, M.U.; Mohtasheemul Hasan, M. Pharmacognostic and phytochemical study of the flowers of Cordia sebestena L. Pak. J. Pharm. Sci. 2022, 35, 69–76. [Google Scholar] [PubMed]
- Chambon, M.; Ho, R.; Baghdikian, B.; Herbette, G.; Bun-Llopet, S.-S.; Garayev, E.; Raharivelomanana, P. Identification of atioxidant metabolites from five plants (Calophyllum inophyllum, Gardenia taitensis, Curcuma longa, Cordia subcordata, Ficus prolixa) of the polynesian pharmacopoeia and cosmetopoeia for skin care. Antioxidants 2023, 12, 1870. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Klessig, D.F. MAPK cascades in plant defense signaling. Trends Plant Sci. 2001, 6, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Z.; Zhang, S.Q. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Bigeard, J.; Hirt, H. Nuclear signaling of plant MAPKs. Front. Plant Sci. 2018, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.X.; Zhu, G.Q.; Liu, Q.; Chen, L.; Li, Y.J.; Hou, B.K. Modulation of plant salicylic acid-associated immune iesponses via glycosylation of dihydroxybenzoic acids. Plant Physiol. 2018, 176, 3103–3119. [Google Scholar] [CrossRef]
- Pedley, K.F.; Martin, G.B. Role of mitogen-activated protein kinases in plant immunity. Curr. Opin. Plant Biol. 2005, 8, 541–547. [Google Scholar] [CrossRef]
- Mithöfer, A.; Maffei, M.E. General mechanisms of plant defense and plant Toxins. In Plant Toxins; Carlini, C., Ligabue-Braun, R., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 3–24. [Google Scholar]
- Sood, M.; Kapoor, D.; Kumar, V.; Kalia, N.; Bhardwaj, R.; Sidhu, G.P.S.; Sharma, A. Mechanisms of plant defense under pathogen stress: A review. Curr. Protein Pept. Sci. 2021, 22, 376–395. [Google Scholar] [CrossRef]
- Kaur, S.; Samota, M.K.; Choudhary, M.; Choudhary, M.; Pandey, A.K.; Sharma, A.; Thakur, J. How do plants defend themselves against pathogens-Biochemical mechanisms and genetic interventions. Physiol. Mol. Biol. Plants 2022, 28, 485–504. [Google Scholar] [CrossRef]
- Liang, P.; Liu, S.; Xu, F.; Jiang, S.; Yan, J.; He, Q.; Liu, W.; Lin, C.; Zheng, F.; Wang, X.; et al. Powdery Mildews are characterized by contracted carbohydrate metabolism and diverse effectors to adapt to obligate biotrophic lifestyle. Front. Microbiol. 2018, 9, 3160. [Google Scholar] [CrossRef]
- Sharma, V.; Hecker, N.; Roscito, J.G.; Foerster, L.; Langer, B.E.; Hiller, M. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat. Commun. 2018, 9, 1215. [Google Scholar] [CrossRef]
- Zhang, T.; Qiao, Q.; Novikova, Y.P.; Wang, Q.; Yue, J.; Guan, Y.; Ming, S.; Liu, T.; De, J.; Liu, Y.; et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. USA 2019, 116, 7137–7146. [Google Scholar] [CrossRef] [PubMed]
- NextDenovo v1.0. Available online: https://github.com/Nextomics/NextDenovo (accessed on 28 May 2020).
- Smartdenovo: Ultra-Fast de Novo Assembler Using Long Noisy Reads. Available online: https://github.com/ruanjue/smartdenovo (accessed on 28 May 2020).
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 2016, 7, 1350. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic. Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic. Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic. Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinf. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Bedell, J.A.; Korf, I.; Gish, W. MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics 2000, 16, 1040–1041. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. Available online: http://www.repeatmasker.org (accessed on 28 May 2020).
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 2019, 1962, 161–177. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Haas, B. TransposonPSI: An Application of PSI-Blast to Mine (Retro-)Transposon ORF Homologies. Available online: https://transposonpsi.sourceforge.net (accessed on 28 May 2020).
- NR: Non-Redundant Protein Sequence Database. Available online: https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins (accessed on 28 June 2020).
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2004, 19, II215–II225. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Sanderson, M.J. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach. Mol. Biol. Evol. 2012, 19, 101–109. [Google Scholar] [CrossRef]
- Smith, S.A.; O’Meara, B.C. treePL: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Zwaenepoel, A.; Van de Peer, Y. WGD-simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics 2019, 35, 2153–2155. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Silva, F.; Van de Peer, Y. Doubletrouble: Identification and Classification of Duplicated Genes. R Package Version 0.99.1. 2022. Available online: https://github.com/almeidasilvaf/doubletrouble (accessed on 21 July 2023).



{kind=link}
{kind=link}
| Type | Contig Length (bp) | Contig Number |
|---|---|---|
| N50 | 16,399,519 | 10 |
| N60 | 15,666,772 | 13 |
| N70 | 14,697,627 | 16 |
| N80 | 12,626,804 | 19 |
| N90 | 10,091,180 | 23 |
| Longest | 42,352,411 | 1 |
| Total | 475,321,542 | 92 |
| Type | Number | Percent (%) |
|---|---|---|
| Complete BUSCOs (C) | 1372 | 95.28 |
| Complete and single-copy BUSCOs (S) | 1303 | 90.49 |
| Complete and duplicated BUSCOs (D) | 69 | 4.79 |
| Fragmented BUSCOs (F) | 14 | 0.97 |
| Missing BUSCOs (M) | 54 | 3.75 |
| Total BUSCO groups searched | 1440 | 100 |
| Databases | Number | Percent (%) |
|---|---|---|
| Swiss-Prot | 21,703 | 81.54 |
| KEGG | 9981 | 37.50 |
| KOG | 14,029 | 52.71 |
| GO | 16,046 | 60.29 |
| NR | 25,549 | 95.99 |
| Total | 25,730 | 96.67 |
| Sequencing Project | Sequencing Platform | Sequence Type | Instrument |
|---|---|---|---|
| Short whole genome sequencing | BGISEQ | PE150 | DNBSEQ-T7 |
| Long whole genome sequencing | OXFORD NANOPORE | Long reads | GridION |
| Short-length transcriptome | BGISEQ | PE150 | DNBSEQ-T7 |
| Full-length transcriptome | BGISEQ | PE150 | DNBSEQ-T7 |
| Hi-C | BGISEQ | PE150 | DNBSEQ-T7 |
| Species Pair | Taxon 1 | Taxon 2 | Estimated Divergent Time (Million Years Ago, MYA) |
|---|---|---|---|
| 1 | Phtheirospermum japonicum | Striga asiatica | 39–64 |
| 2 | Handroanthus impetiginosus | Sesamum indicum | 56–73 |
| 3 | Coffea eugenioides | Cordia subcordata | 75–96 |
| 4 | Capsicum chinense | Solanum tuberosum | 18.2–31.4 |
| 5 | Rhododendron simsii | Nyssa sinensis | 105–119 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-L.; Wang, Z.-F.; Jian, S.-G.; Liao, H.-M.; Liu, D.-M. Genome Assembly of Cordia subcordata, a Coastal Protection Species in Tropical Coral Islands. Int. J. Mol. Sci. 2023, 24, 16273. https://doi.org/10.3390/ijms242216273
Chen Y-L, Wang Z-F, Jian S-G, Liao H-M, Liu D-M. Genome Assembly of Cordia subcordata, a Coastal Protection Species in Tropical Coral Islands. International Journal of Molecular Sciences. 2023; 24(22):16273. https://doi.org/10.3390/ijms242216273
Chicago/Turabian StyleChen, Yi-Lan, Zheng-Feng Wang, Shu-Guang Jian, Hai-Min Liao, and Dong-Ming Liu. 2023. "Genome Assembly of Cordia subcordata, a Coastal Protection Species in Tropical Coral Islands" International Journal of Molecular Sciences 24, no. 22: 16273. https://doi.org/10.3390/ijms242216273