
Current Progress, Applications and Challenges of Multi-Omics Approaches in Sesame Genetic Improvement
 ,
,  ,
,  and
and 
Abstract
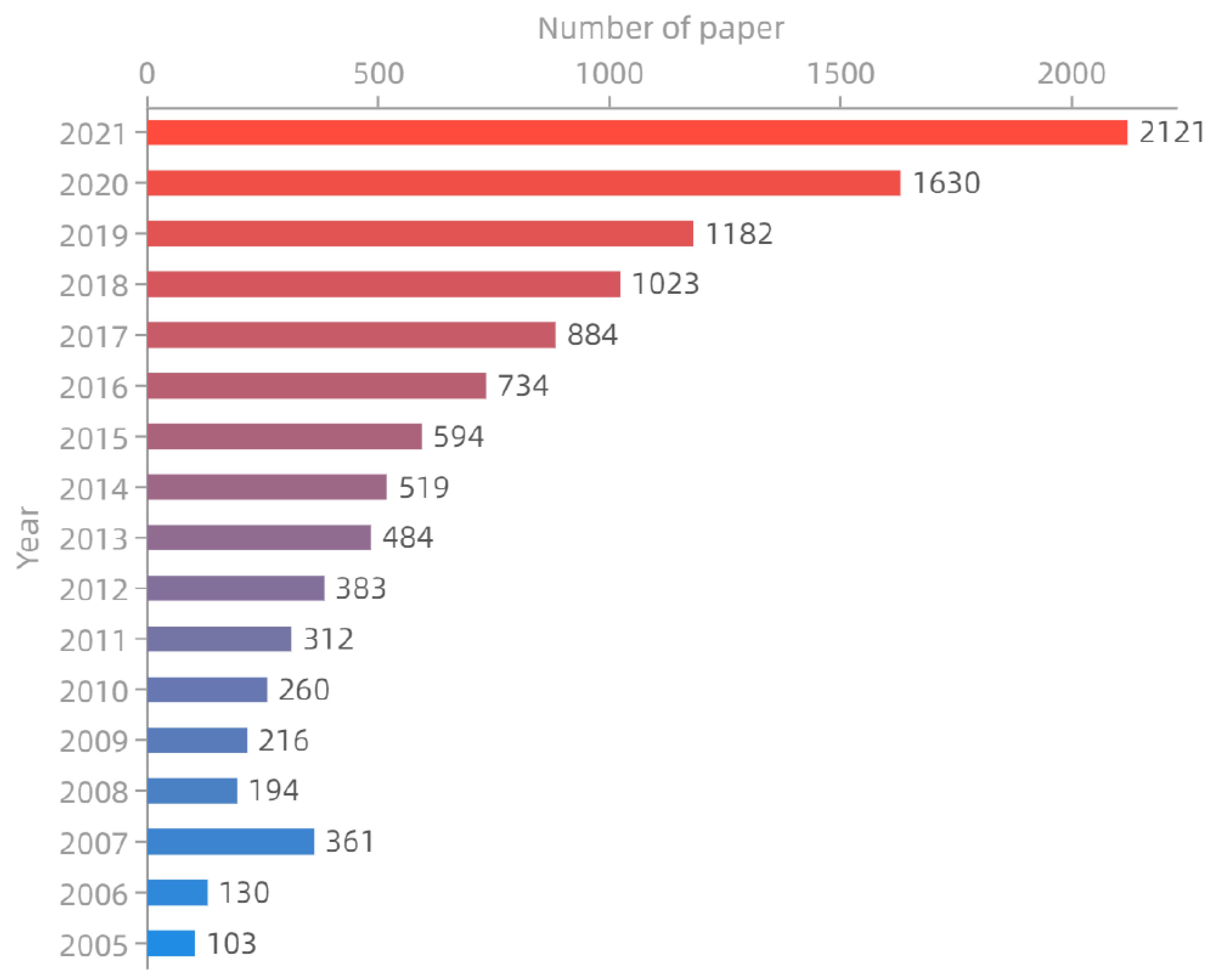
:1. Introduction
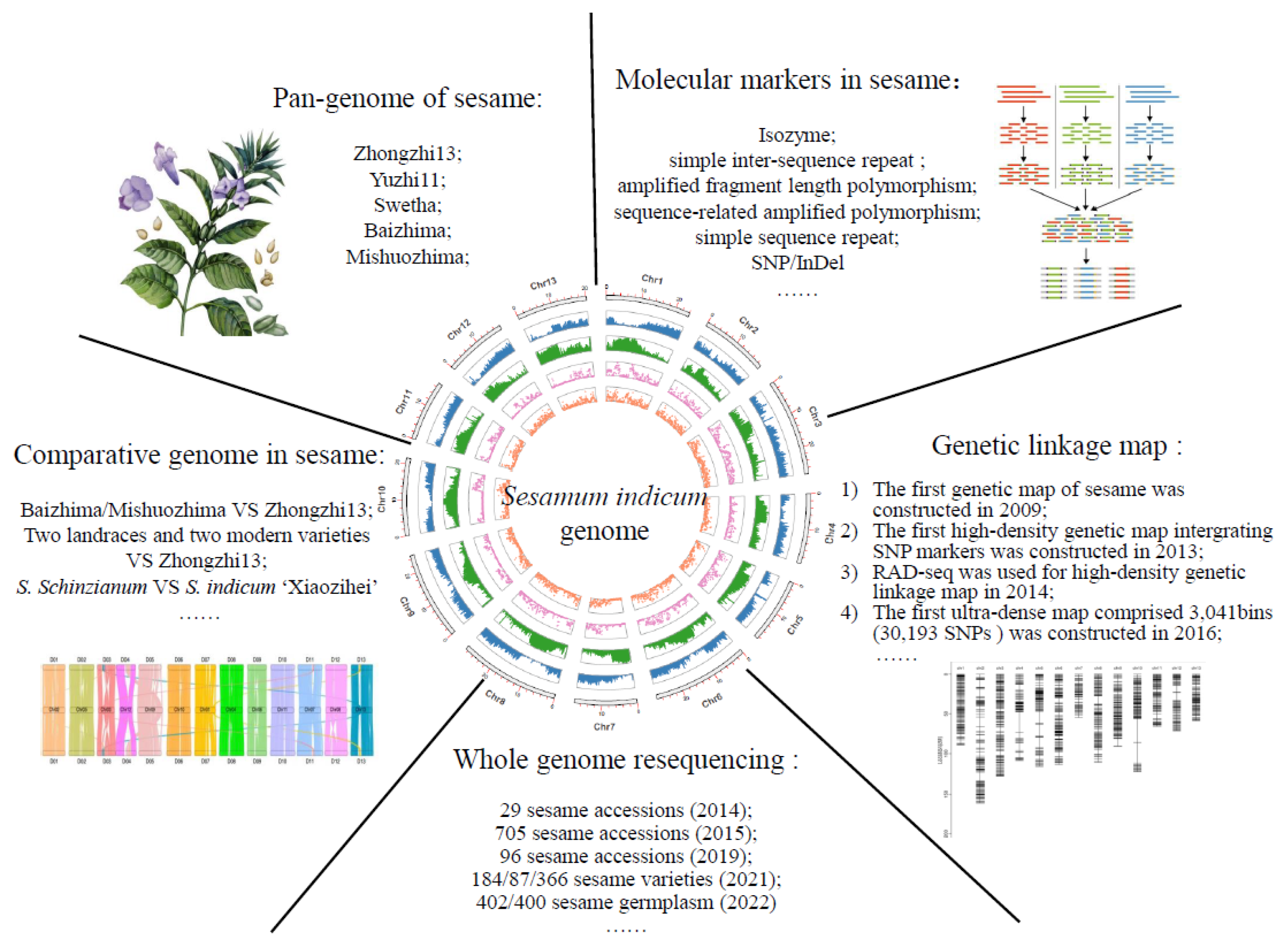
2. Genomics
2.1. Genome Sequencing in Sesame
2.1.1. The Nuclear Genome in Sesame
2.1.2. The Nuclear Genome in Sesame
2.2. Applications of Sesame Genomics
2.2.1. Molecular Marker in Sesame
2.2.2. High-Density Genetic Linkage Map of Sesame
2.2.3. Whole Genome Resequencing in Sesame
2.2.4. The Application of Comparative Genomics in Sesame
2.2.5. The Pan-Genome of Sesame
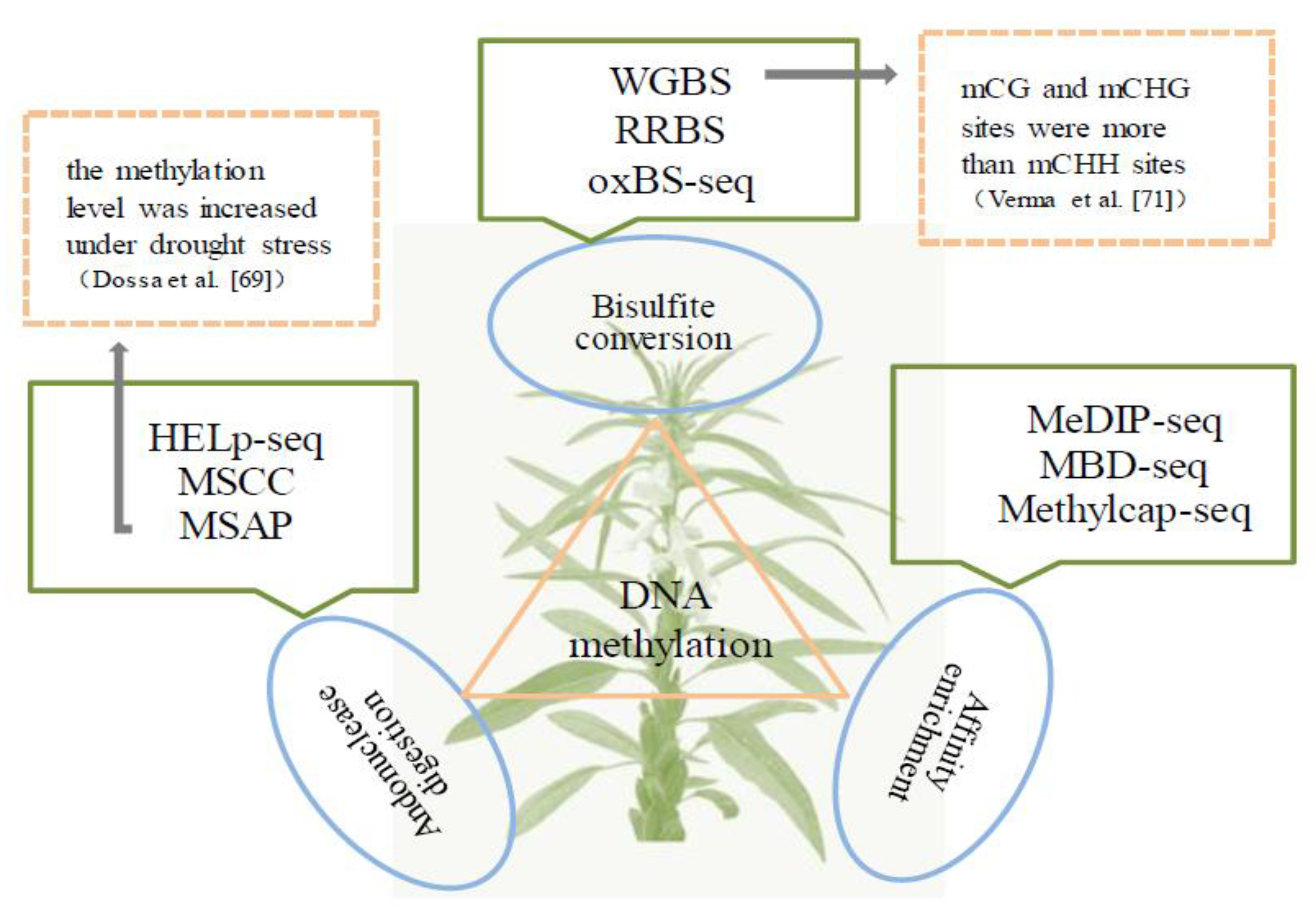
3. Methylomics
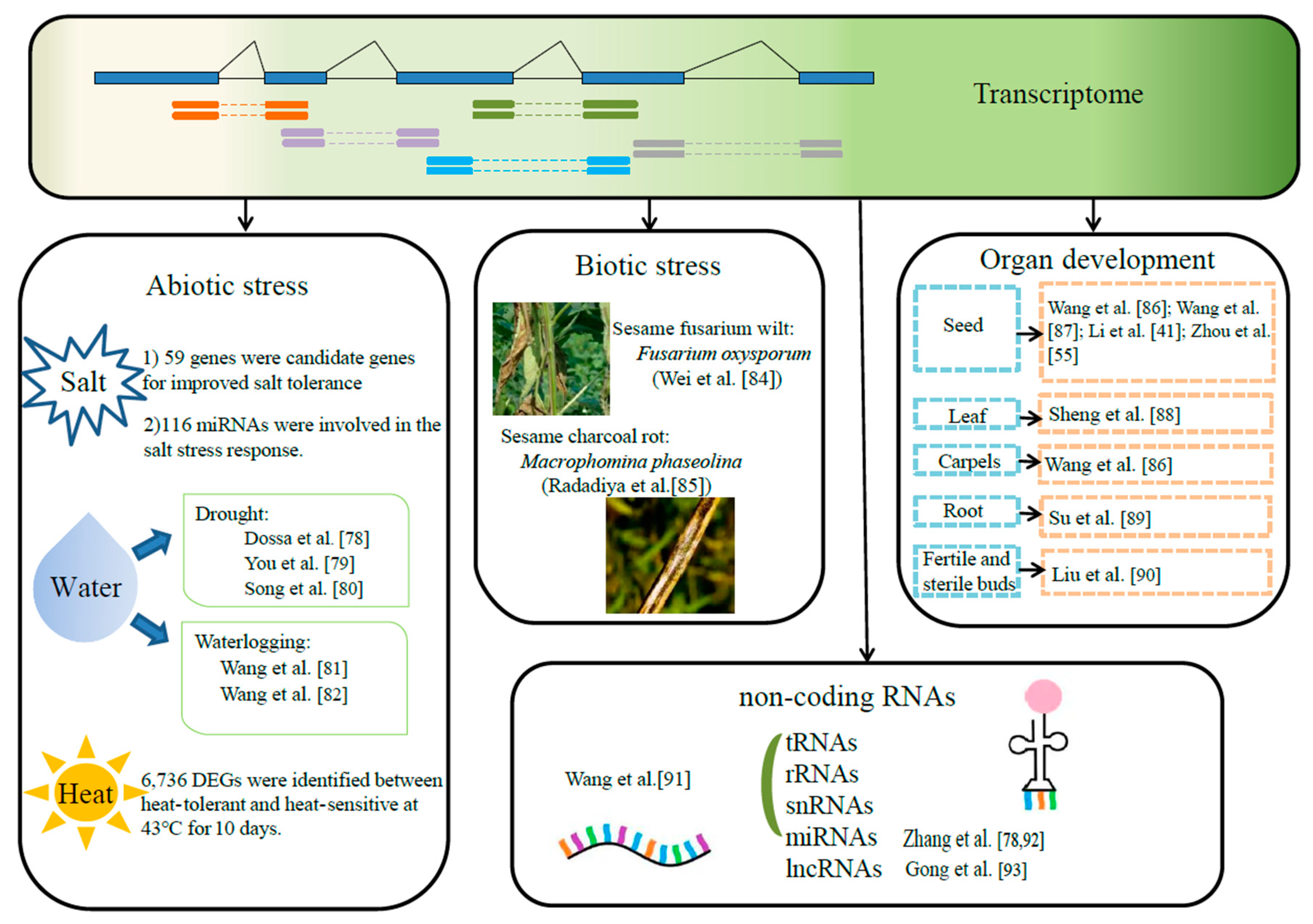
4. Transcriptomics
4.1. Application of Transcriptomics in Abiotic Stress
4.1.1. Salt Stress
4.1.2. Water Stress
4.1.3. Heat Stress
4.2. Application of Transcriptomics in Biotic Stress
4.3. Application of Transcriptomics in Organ Development
4.4. Research on Non-Coding RNAs
5. Proteomics
6. Metabolomics
6.1. Research on Abiotic Stress
6.2. Research on Important Traits
7. The Challenges of OMICS Approaches for Sesame Genetic Improvements
- (1)
- Strengthen the development of markers related to the key agronomic traits of sesame: there are few markers related to important agronomic traits (such as resistance, yield and quality), which greatly limits the application of molecular marker-assisted selection in breeding. Therefore, we should excavate accurately and efficiently the markers that are closely linked to complex agronomic traits from several aspects: in-depth excavation of the genomic variation to obtain structure variation materials; improvement of the algorithm of GWAS or QTL to increase the detection force, such as a multi-sites GWAS method for detecting the rare sites. In addition, the existing molecular markers have a poor stability and low genetic effect in breeding. We need to improve the technology to really apply the effective molecular markers in sesame breeding.
- (2)
- Analyzing the genetic mechanism of complex agronomic traits based on multi-omics: by combining the data of genomics, transcriptome, proteome and metabolomics, it the regulatory genes of complex agronomic traits could be revealed, their mechanism of actions, regulatory network and metabolic pathways could be clarified, and a theoretical basis and gene resources for modern molecular breeding (transgenic or gene editing) could be provided.
- (3)
- Tightening modern biotechnology research and combining it with conventional breeding: the research of sesame cell engineering technology should be further strengthened to make it widely used in sesame breeding practice. The study of molecular marker-assisted selection breeding technology system should be further enhanced, and the obtained markers should be gradually applied to breeding practice. We should tighten the research on the transgenic technology of sesame disease resistance and stress-resistant genes, as well as the research on the heredity and safety evaluation of transgenic plant traits. In addition, we should strengthen the combination of cell engineering breeding, molecular breeding and conventional breeding, pay attention to the research of basic breeding theory and efficient breeding technology, and gradually move towards molecular design breeding.
8. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed gene |
| IR | Inverted repeats |
| LSC | Large single copy |
| SSC | Small single copy |
| ISSR | Simple inter-sequence repeat |
| AFLR | Amplified fragment length polymorphism |
| SRAP | Sequence-related amplified polymorphism |
| SSRS | Simple sequence repeats |
| SNP | Single nucleotide polymorphism |
| INDEL | Insertion/deletion |
| LG | Linkage groups |
| SLAF-SEQ | Specific length amplified fragment sequencing |
| RAD-SEQ | Restriction-site associated DNA sequencing |
| GBS | Genotyping by sequencing |
| WGS | Whole-genome resequencing |
| WGBS | Whole genome bisulfite methylation sequencing |
| OXBS-SEQ | Oxidative bisulfite sequencing |
| RPBS | Reduced representation bisulfite sequencing |
| MEDIP-SEQ | Methylation DNA immunoprecipitation sequencing |
| HELP-SEQ | HpaⅡtiny fragement enrichment by ligation-mediated PCR |
| HPI | Hour post inoculation |
| DPA | Days post-anthesis |
| DAP | Differential abundant proteins |
References
- Biswas, S.; Natta, S.; Ray, D.P.; Mondal, P.; Saha, U. Til (Sesamum indicum L.)-An underexploited but promising Oilseed with multifarious applications: A Review. Int. J. Bioresource Sci. 2018, 5, 127–139. [Google Scholar] [CrossRef]
- Rangkadilok, N.; Pholphana, N.; Mahidol, C.; Wongyai, W.; Saengsooksree, K.; Nookabkaew, S.; Satayavivad, J. Variation of sesamin, sesamolin and tocopherols in sesame (Sesamum indicum L.) seeds and oil products in Thailand. Food Chem. 2010, 122, 724–730. [Google Scholar] [CrossRef]
- Witcombe, J.R.; Hollington, P.A.; Howarth, C.J.; Reader, S.; Steele, K.A. Breeding for abiotic stresses for sustainable agriculture. Philos. Trans. R. Soc. B 2008, 363, 703–716. [Google Scholar] [CrossRef]
- Wang, L.; Yu, S.; Tong, C.; Zhao, Y.; Liu, Y.; Song, C.; Zhang, Y.; Zhang, X.; Wang, Y.; Hua, W.; et al. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Boil. 2014, 15, R39. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Liu, H.; Wang, X.; Li, W.; Chen, M.S.; Niu, L.J. Chromatin architectures are associated with response to dark treatment in the oil crop Sesamum indicum, based on a high-quality genome assembly. Plant Cell Physiol. 2020, 61, 978–987. [Google Scholar] [CrossRef]
- Yu, J.; Golicz, A.A.; Lu, K.; Dossa, K.; Zhang, Y.; Chen, J.; Wang, L.; You, J.; Fan, D.; Edwards, D.; et al. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 2019, 17, 881–892. [Google Scholar] [CrossRef]
- Wang, M.; Huang, J.; Liu, S.; Liu, X.; Li, R.; Luo, J.; Fu, Z. Improved assembly and annotation of the sesame genome. DNA Res. 2022, 29, dsac041. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Lin, Q.; Lu, J.; Lv, S.; Zhang, Y.; Wang, X.; Fan, W.; Liu, W.; Zhang, L.; et al. The wild allotetraploid sesame genome provides novel insights into evolution and lignan biosynthesis. J. Adv. Res. 2022. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Olejniczak, S.A.; Łojewska, E.; Kowalczyk, T.; Sakowicz, T. Chloroplasts: State of research and practical applications of plastome sequencing. Planta 2016, 244, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Yi, D.K.; Kim, K.J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PloS ONE 2013, 8, e80508. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, B.; Collard, B.C.; Demont, M. Improving global food security through accelerated plant breeding. Plant Sci. 2019, 287, 110207. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Layrisse, A. Isozyme resemblance among forty sesame accessions. Sesame Safflower Newsl. 2001, 16, 32–35. [Google Scholar]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Cri. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Kumar, H.; Kaur, G.; Banga, S. Molecular characterization and assessment of genetic diversity in sesame (Sesamum indicum L.) germplasm collection using ISSR markers. J. Crop Improv. 2012, 26, 540–557. [Google Scholar] [CrossRef]
- Frary, A.; Tekin, P.; Celik, I.; Furat, S.; Uzun, B.; Doganlar, S. Morphological and molecular diversity in Turkish sesame germplasm and core set selection. Crop Sci. 2015, 55, 702–711. [Google Scholar] [CrossRef]
- Ali AL-somain, B.H.; Migdadi, H.M.; Al-Faifi, S.A.; Alghamdi, S.S.; Muharram, A.A.; Mohammed, N.A.; Refay, Y.A. Assessment of genetic diversity of sesame accessions collected from different ecological regions using sequence-related amplified polymorphism markers. 3 Biotech 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Adu-Gyamfi, R.; Prempeh, R.; Zakaria, I. Diversity assessment of some sesame (Sesamum indicum L.) genotypes cultivated in Northern Ghana using morphological and simple sequence repeat (SSR) markers. Adv. Agric. 2019, 2019, 6067891. [Google Scholar]
- Wei, L.; Miao, H.; Li, C.; Duan, Y.; Niu, J.; Zhang, T.; Zhao, Q.; Zhang, H. Development of SNP and InDel markers via de novo transcriptome assembly in Sesamum indicum L. Mol. Breed. 2014, 34, 2205–2217. [Google Scholar] [CrossRef]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Basak, M.; Uzun, B.; Yol, E. Genetic diversity and population structure of the Mediterranean sesame core collection with use of genome-wide SNPs developed by double digest RAD-Seq. PLoS ONE 2019, 14, e0223757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudhana, R. Linkage Mapping. In Sorghum Molecular Breeding; Springer: New Delhi, India, 2015; pp. 47–70. [Google Scholar]
- Wei, L.B.; Zhang, H.Y.; Zheng, Y.Z.; Miao, H.M.; Zhang, T.Z.; Guo, W.Z. A genetic linkage map construction for sesame (Sesamum indicum L.). Genes Genom. 2009, 31, 199–208. [Google Scholar] [CrossRef]
- Zhang, H.; Miao, H.; Wei, L.; Li, C.; Zhao, R.; Wang, C. Genetic analysis and QTL mapping of seed coat color in sesame (Sesamum indicum L.). PloS ONE 2013, 8, e63898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Li, D.; Gao, Y.; Lu, H.; Zhang, X. Mapping of sesame waterlogging tolerance QTL and identification of excellent waterlogging tolerant germplasm. Sci. Agric. Sin. 2014, 47, 422–430. [Google Scholar]
- Wang, L.; Zhang, Y.; Zhu, X.; Zhu, X.; Li, D.; Zhang, X.; Gao, Y.; Xiao, G.; Wei, X.; Zhang, X. Development of an SSR-based genetic map in sesame and identification of quantitative trait loci associated with charcoal rot resistance. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Xu, F.; Zhou, R.; Dossou, S.S.K.; Song, S.; Wang, L. Fine mapping of a major pleiotropic QTL associated with sesamin and sesamolin variation in sesame (Sesamum indicum L.). Plants 2021, 10, 1343. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Xin, H.; Li, D.; Ma, C.; Ding, X.; Hong, W.; Zhang, X. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 2013, 13, 1–12. [Google Scholar] [CrossRef]
- Du, H.; Zhang, H.; Wei, L.; Li, C.; Duan, Y.; Wang, H. A high-density genetic map constructed using specific length amplified fragment (SLAF) sequencing and QTL mapping of seed-related traits in sesame (Sesamum indicum L.). BMC Plant Biol. 2019, 19, 1–20. [Google Scholar] [CrossRef]
- Asekova, S.; Oh, E.; Kulkarni, K.P.; Siddique, M.I.; Lee, M.H.; Kim, J.I.; Lee, J.D.; Kim, M.; Oh, K.W.; Ha, T.J.; et al. An integrated approach of QTL mapping and genome-wide association analysis identifies candidate genes for phytophthora blight resistance in sesame (Sesamum indicum L.). Front. Plant Sci. 2021, 12, 604709. [Google Scholar] [CrossRef]
- Mei, H.; Liu, Y.; Cui, C.; Hu, C.; Xie, F.; Zheng, L.; Du, Z.; Wu, K.; Jiang, X.; Zheng, Y.; et al. QTL mapping of yield-related traits in sesame. Mol. Breed. 2021, 41, 1–14. [Google Scholar] [CrossRef]
- Yol, E.; Basak, M.; Kızıl, S.; Lucas, S.J.; Uzun, B. A high-density SNP genetic map construction using ddRAD-Seq and mapping of capsule shattering trait in sesame. Front. Plant Sci. 2021, 12, 679659. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, Q.; Zhang, Y.; Zhu, X.; Zhu, X.; Li, D.; Ni, X.; Gao, Y.; Xiang, H.; Wei, X.; et al. Updated sesame genome assembly and fine mapping of plant height and seed coat color QTLs using a new high-density genetic map. BMC Genom. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uncu, A.O.; Frary, A.; Karlovsky, P.; Doganlar, S. High-throughput single nucleotide polymorphism (SNP) identification and mapping in the sesame (Sesamum indicum L.) genome with genotyping by sequencing (GBS) analysis. Mol. Breed. 2016, 36, 1–12. [Google Scholar] [CrossRef]
- Mei, H.; Liu, Y.; Du, Z.; Wu, K.; Cui, C.; Jiang, X.; Zhang, H.; Zheng, Y. High-density genetic map construction and gene mapping of basal branching habit and flowers per leaf axil in sesame. Front. Plant Sci. 2017, 8, 636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Miao, H.; Li, C.; Wei, L.; Duan, Y.; Ma, Q.; Kong, J.; Xu, F.; Chang, S. Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Sci. Rep. 2016, 6, 31556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Miao, H.; Wei, L.; Li, C.; Duan, Y.; Xu, F.; Qu, W.; Zhao, R.; Ju, M.; Chang, S. Identification of a SiCL1 gene controlling leaf curling and capsule indehiscence in sesame via cross-population association mapping and genomic variants screening. BMC Plant Biol. 2018, 18, 1–12. [Google Scholar] [CrossRef]
- Teboul, N.; Gadri, Y.; Berkovich, Z.; Reifen, R.; Peleg, Z. Genetic architecture underpinning yield components and seed mineral-nutrients in sesame. Genes 2020, 11, 1221. [Google Scholar] [CrossRef]
- Liang, J.; Sun, J.; Ye, Y.; Yan, X.; Yan, T.; Rao, Y.; Zhou, H.; Le, M. QTL mapping of PEG-induced drought tolerance at the early seedling stage in sesame using whole genome re-sequencing. PLoS ONE 2021, 16, e0247681. [Google Scholar] [CrossRef]
- Li, C.; Duan, Y.; Miao, H.; Ju, M.; Wei, L.; Zhang, H. Identification of candidate genes regulating the seed coat color trait in sesame (Sesamum indicum L.) using an integrated approach of QTL mapping and transcriptome analysis. Front. Genet. 2021, 12, 1369. [Google Scholar] [CrossRef]
- Fuentes-Pardo, A.P.; Ruzzante, D.E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef]
- Wang, L.; Han, X.; Zhang, Y.; Li, D.; Wei, X.; Ding, X.; Zhang, X. Deep resequencing reveals allelic variation in Sesamum indicum. BMC Plant Biol. 2014, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, K.; Zhang, Y.; Feng, Q.; Wang, L.; Zhao, Y.; Li, D.; Zhao, Q.; Zhu, X.; Zhu, X.; et al. Genetic discovery for oil production and quality in sesame. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Dossa, K.; Zhang, Y.; Wei, X.; Wang, L.; Zhang, Y.; Liu, A.; Zhou, R.; Zhang, X. GWAS uncovers differential genetic bases for drought and salt tolerances in sesame at the germination stage. Genes 2018, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Dossa, K.; Li, D.; Zhou, R.; Yu, J.; Wang, L.; Zhang, Y.; You, J.; Liu, A.; Mmadi, M.; Fonceka, D.; et al. The genetic basis of drought tolerance in the high oil crop Sesamum indicum. Plant Biotechnol. J. 2019, 17, 1788–1803. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Dossa, K.; Li, D.; Yu, J.; You, J.; Wei, X.; Zhang, X. Genome-wide association studies of 39 seed yield-related traits in sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2018, 19, 2794. [Google Scholar] [CrossRef]
- Dossa, K.; Zhou, R.; Li, D.; Liu, A.; Qin, L.; Mmadi, M.A.; Su, R.; Zhang, Y.; Wang, J.; Gao, Y.; et al. A novel motif in the 5’-UTR of an orphan gene ‘Big Root Biomass’ modulates root biomass in sesame. Plant Biotechnol. J. 2021, 19, 1065–1079. [Google Scholar] [CrossRef]
- Wang, X.; You, J.; Liu, A.; Qi, X.; Li, D.; Zhao, Y.; Zhang, Y.; Zhang, L.; Zhang, X.; Li, P. Variation in melatonin contents and genetic dissection of melatonin biosynthesis in sesame. Plants 2022, 11, 2005. [Google Scholar] [CrossRef]
- Song, S.; Zhang, L.; Zhao, Y.; Sheng, C.; Zhou, W.; Dossou, S.S.K.; Wang, L.; You, J.; Zhou, R.; Wei, X.; et al. Metabolome genome-wide association study provides biochemical and genetic insights into natural variation of primary metabolites in sesame. Plant J. 2022, 112, 1051–1069. [Google Scholar] [CrossRef]
- He, Q.; Xu, F.; Min, M.H.; Chu, S.H.; Kim, K.W.; Park, Y.J. Genome-wide association study of vitamin E using genotyping by sequencing in sesame (Sesamum indicum). Genes Genom. 2019, 41, 1085–1093. [Google Scholar] [CrossRef]
- Sabag, I.; Morota, G.; Peleg, Z. Genome-wide association analysis uncovers the genetic architecture of tradeoff between flowering date and yield components in sesame. BMC Plant Biol. 2021, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Liu, Y.; Liu, Y.; Cui, X.; Sun, Z.; Du, Z.; Wu, K.; Jiang, X.; Mei, H.; Zheng, Y. Genome-wide association study of seed coat color in sesame (Sesamum indicum L.). PLoS ONE 2021, 16, e0251526. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Q.; Dossou, S.S.K.; Zhou, R.; Zhao, Y.; Zhou, W.; Zhang, Y.; Li, D.; You, J.; Wang, L. Genome-wide association study uncovers loci and candidate genes underlying phytosterol variation in sesame (Sesamum indicum L.). Agricalture 2022, 12, 392. [Google Scholar] [CrossRef]
- Zhou, W.; Song, S.; Dossou, S.S.K.; Zhou, R.; Wei, X.; Wang, Z.; Sheng, C.; Zhang, Y.; You, J.; Wang, L. Genome-wide association analysis and transcriptome reveal novel loci and a candidate regulatory gene of fatty acid biosynthesis in sesame (Sesamum indicum L.). Plant Physiol. Bioch. 2022, 186, 220–231. [Google Scholar] [CrossRef]
- Dossou, S.S.K.; Song, S.; Liu, A.; Li, D.; Zhou, R.; Berhe, M.; Zhang, Y.; Sheng, C.; Wang, Z.; You, J.; et al. Resequencing of 410 sesame accessions identifies SINST1 as the major underlying gene for lignans variation. Int. J. Mol. Sci. 2023, 24, 1055. [Google Scholar] [CrossRef]
- Berhe, M.; Dossa, K.; You, J.; Mboup, P.A.; Diallo, I.N.; Diouf, D.; Zhang, X.; Wang, L. Genome-wide association study and its applications in the non-model crop Sesamum indicum. BMC Plant Biol. 2021, 21, 1–19. [Google Scholar] [CrossRef]
- Wei, X.; Zhu, X.; Yu, J.; Wang, L.; Zhang, Y.; Li, D.; Zhou, R.; Ju, M.; Zhang, X. Identification of sesame genomic variations from genome comparison of landrace and variety. Front. Plant Sci. 2016, 7, 1169. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Morgante, M.; De Paoli, E.; Radovic, S. Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 1–16. [Google Scholar]
- Della Coletta, R.; Qiu, Y.; Ou, S.; Hufford, M.B.; Hirsch, C.N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 2021, 22, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulos, C.; Giehr, P.; Wolf, V. H(O)TA: Estimation of DNA methylation and hydroxylation levels and efficiencies from time course data. Bioinformatics 2017, 33, 1733–1734. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [Green Version]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef]
- Oda, M.; Glass, J.L.; Thompson, R.F.; Mo, Y.; Olivier, E.N.; Figueroa, M.E.; Selzer, R.R.; Richmond, T.A.; Zhang, X.; Dannenberg, L.; et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 2009, 37, 3829–3839. [Google Scholar] [CrossRef]
- Rehman, M.; Tanti, B. Understanding epigenetic modifications in response to abiotic stresses in plants. Biocatal. Agric. Biotechnol. 2020, 27, 101673. [Google Scholar] [CrossRef]
- Dossa, K.; Ali Mmadi, M.; Zhou, R.; Zhou, Q.; Yang, M.; Cisse, N.; Diouf, D.; Wang, L.; Xiurong, Z. The contrasting response to drought and waterlogging is underpinned by divergent DNA methylation programs associated with transcript accumulation in sesame. Plant Sci. 2018, 277, 207–217. [Google Scholar]
- Singh, A.; Verma, P.; Lakhanpaul, S. Exploring the methylation status of selected flowering genes in healthy and phyllody infected sesame plants. Phytopathogenic Mollicutes 2020, 10, 36–42. [Google Scholar] [CrossRef]
- Verma, P.; Singh, A.; Purru, S.; Bhat, K.V.; Lakhanpaul, S. Comparative DNA methylome of phytoplasma associated retrograde metamorphosis in sesame (Sesamum indicum L.). Biology 2022, 11, 954. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Messing, J.; Bharti, A.K.; Karlowski, W.M.; Gundlach, H.; Kim, H.R.; Yu, Y.; Wei, F.; Fuks, G.; Soderlund, C.A.; Mayer, K.F.X.; et al. Sequence composition and genome organization of maize. Proc. Natl. Acad. Sci. USA 2004, 101, 14349–14354. [Google Scholar] [CrossRef] [PubMed]
- Barrell, P.J.; Meiyalaghan, S.; Jacobs, J.M.; Conner, A.J. Applications of biotechnology and genomics in potato improvement. Plant Biotechnol. J. 2013, 11, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Feuillet, C.; Leach, J.E.; Rogers, J.; Schnable, P.S.; Eversole, K. Crop genome sequencing: Lessons and rationales. Trends Plant Sci. 2011, 16, 77–88. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhou, R.; Wang, X.; Dossa, K.; Wang, L.; Zhang, Y.; Yu, J.; Gong, H.; Zhang, X.; et al. Transcriptome and metabolome analyses of two contrasting sesame genotypes reveal the crucial biological pathways involved in rapid adaptive response to salt stress. BMC Plant Biol. 2019, 19, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gong, H.; Li, D.; Zhou, R.; Zhao, F.; Zhang, X.; You, J. Integrated small RNA and degradome sequencing provide insights into salt tolerance in sesame (Sesamum indicum L.). BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Dossa, K.; Li, D.; Wang, L.; Zheng, X.; Liu, A.; Yu, J.; Wei, X.; Zhou, R.; Fonceka, D.; Diouf, D.; et al. Transcriptomic, biochemical and physio-anatomical investigations shed more light on responses to drought stress in two contrasting sesame genotypes. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- You, J.; Zhang, Y.; Liu, A.; Li, D.; Wang, X.; Dossa, K.; Zhou, Z.; Yu, J.; Zhang, Y.; Wang, L.; et al. Transcriptomic and metabolomic profiling of drought-tolerant and susceptible sesame genotypes in response to drought stress. BMC Plant Biol. 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Song, Q.; Joshi, M.; Wang, S.; Johnson, C.D.; Joshi, V. Comparative analysis of root transcriptome profiles of sesame (Sesamum indicum L.) in response to osmotic stress. J. Plant Growth Regul. 2021, 40, 1787–1801. [Google Scholar] [CrossRef]
- Wang, L.; Li, D.; Zhang, Y.; Gao, Y.; Yu, J.; Wei, X.; Zhang, X. Tolerant and susceptible sesame genotypes reveal waterlogging stress response patterns. PLoS ONE 2016, 11, e0149912. [Google Scholar] [CrossRef]
- Wang, L.; Dossa, K.; You, J.; Zhang, Y.; Li, D.; Zhou, R.; Yu, J.; Wei, X.; Zhu, X.; Gao, Y.; et al. High-resolution temporal transcriptome sequencing unravels ERF and WRKY as the master players in the regulatory networks underlying sesame responses to waterlogging and recovery. Genomics 2021, 113, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gao, T.; Zhang, P.; Li, F.; Wang, D.; Tian, Y.; Lu, H.; Zhang, H.; Wei, S. Comparative physiological and transcriptomic analysis of sesame cultivars with different tolerance responses to heat stress. Physiol. Mol. Biol. Pla. 2022, 28, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhang, H.; Duan, Y.; Li, C.; Chang, S.; Miao, H. Transcriptome comparison of resistant and susceptible sesame (Sesamum indicum L.) varieties inoculated with Fusarium oxysporum f. sp. Sesame. Plant Breed. 2016, 135, 627–635. [Google Scholar] [CrossRef]
- Radadiya, N.; Mangukia, N.; Antala, V.; Desai, H.; Chaudhari, H.; Dholaria, T.L.; Dholaria, D.; Tomar, R.S.; Golakiya, B.A.; Mahatma, M.K. Transcriptome analysis of sesame-Macrophomina phaseolina interactions revealing the distinct genetic components for early defense responses. Physiol. Mol. Biol. Pla. 2021, 27, 1675–1693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Li, D.; Dossa, K.; Wang, M.L.; Zhou, R.; Yu, J.; Zhang, X. Gene expression profiles that shape high and low oil content sesames. BMC Genet. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dossou, S.S.K.; Wei, X.; Zhang, Y.; Li, D.; Yu, J.; Zhang, X. Transcriptome dynamics during black and white sesame (Sesamum indicum L.) seed development and identification of candidate genes associated with black pigmentation. Genes 2020, 11, 1399. [Google Scholar] [CrossRef]
- Sheng, C.; Song, S.; Zhou, R.; Li, D.; Gao, Y.; Cui, X.; Tang, X.; Zhang, X.; Tu, J.; Zhang, X.; et al. QTL-seq and transcriptome analysis disclose major QTL and candidate genes controlling leaf size in Sesame (Sesamum indicum L.). Front. Plant Sci. 2021, 12, 580846. [Google Scholar] [CrossRef]
- Su, R.; Zhou, R.; Mmadi, M.A.; Li, D.; Qin, L.; Liu, A.; Wang, J.; Gao, Y.; Wei, M.; Shi, L.; et al. Root diversity in sesame (Sesamum indicum L.): Insights into the morphological, anatomical and gene expression profiles. Planta 2019, 250, 1461–1474. [Google Scholar] [CrossRef]
- Liu, H.; Tan, M.; Yu, H.; Li, L.; Zhou, F.; Yang, M.; Zhou, T.; Zhao, Y. Comparative transcriptome profiling of the fertile and sterile flower buds of a dominant genic male sterile line in sesame (Sesamum indicum L.). BMC Plant Biol. 2016, 16, 1–13. [Google Scholar] [CrossRef]
- Brosnan, C.A.; Voinnet, O. The long and the short of noncoding RNAs. Curr. Opin. Cell Biol. 2009, 21, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Zhang, Y.Y.; Thakur, K.; Zhang, F.; Hu, F.; Zhang, J.G.; Wei, P.C.; Wei, Z.J. Integration of miRNAs, degradome, and transcriptome omics uncovers a complex regulatory network and provides insights into lipid and fatty acid synthesis during sesame seed development. Front. Plant Sci. 2021, 12, 1529. [Google Scholar]
- Gong, H.; You, J.; Zhang, X.; Liu, Y.; Zhao, F.; Cui, X.; Zhang, Y. Genome-wide identification and functional analysis of long non-coding RNAs in sesame response to salt stress. J. Plant Biol. 2021, 64, 555–565. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Verma, N.; Sao, P.; Srivastava, A.; Singh, S. Physiological and Molecular Responses to Drought, Submergence and Excessive Watering in Plants. In Harsh Environment and Plant Resilience; Springer: Cham, Switzerland, 2021; pp. 305–321. [Google Scholar]
- Dossa, K.; You, J.; Wang, L.; Zhang, Y.; Li, D.; Zhou, R.; Yu, J.; Wei, X.; Zhu, X.; Jiang, S.; et al. Transcriptomic profiling of sesame during waterlogging and recovery. Sci. DATA 2019, 6, 1–5. [Google Scholar] [CrossRef]
- Essemine, J.; Ammar, S.; Bouzid, S. Impact of heat stress on germination and growth in higher plants: Physiological, biochemical and molecular repercussions and mechanisms of defence. J. Biol. Sci. 2010, 10, 565–572. [Google Scholar] [CrossRef]
- Elewa, I.S.; Mostafa, M.H.; Sahab, A.F.; Ziedan, E.H. Direct effect of biocontrol agents on wilt and root-rot diseases of sesame. Arch. Phytopathol. Plant Prot. 2011, 44, 493–504. [Google Scholar] [CrossRef]
- Haque, S.; Ahmad, J.S.; Clark, N.M.; Williams, C.M.; Sozzani, R. Computational prediction of gene regulatory networks in plant growth and development. Curr. Opin. Plant Biol. 2019, 47, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Jamalkandi, S.A.; Masoudi-Nejad, A. Reconstruction of Arabidopsis thaliana fully integrated small RNA pathway. Funct. Integr. Genomic. 2009, 9, 419–432. [Google Scholar] [CrossRef]
- Fedak, H.; Palusinska, M.; Krzyczmonik, K.; Brzezniak, L.; Yatusevich, R.; Pietras, Z.; Kaczanowski, S.; Swiezewski, S. Control of seed dormancy in Arabidopsis by a cis-acting noncoding antisense transcript. Proc. Natl. Acad. Sci. USA 2016, 113, E7846–E7855. [Google Scholar] [CrossRef]
- Hossain, Z.; Nouri, M.Z.; Komatsu, S. Plant cell organelle proteomics in response to abiotic stress. J. Proteome Res. 2012, 11, 37–48. [Google Scholar] [CrossRef]
- Jung, H.J.; Roy, S.K.; Cho, S.W.; Kwon, S.J.; Kun, C.; Chun, H.C.; Woo, S.H. Proteome analysis of sesame leaves in response to waterlogging stress at vegetative and flowering stages. Biol. Plant. 2019, 63, 733–749. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, M.; Liu, A.; Zhou, R.; Li, D.; Dossa, K.; Wang, L.; Zhang, Y.; Gong, H.; Zhang, X.; et al. Comparative proteomic analysis of two sesame genotypes with contrasting salinity tolerance in response to salt stress. J. Proteom. 2019, 201, 73–83. [Google Scholar] [CrossRef]
- Pamei, I.; Makandar, R. Comparative proteome analysis reveals the role of negative floral regulators and defense-related genes in phytoplasma infected sesame. Protoplasma 2022, 259, 1441–1453. [Google Scholar] [CrossRef]
- Debnath, M.; Pandey, M.; Bisen, P.S. An omics approach to understand the plant abiotic stress. OMICS 2011, 15, 739–762. [Google Scholar] [CrossRef]
- Saddique, M.; Kamran, M.; Shahbaz, M. Differential Responses of Plants to Biotic Stress and the Role of Metabolites. In Plant Metabolites and Regulation under Environmental Stress; Academic Press: Amsterdam, The Netherlands, 2018; pp. 69–87. [Google Scholar]
- Khare, S.; Singh, N.B.; Singh, A.; Hussain, I.; Niharika, K.; Yadav, V.; Bano, C.; Yadav, R.K.; Amist, N. Plant secondary metabolites synthesis and their regulations under biotic and abiotic constraints. J. Plant Biol. 2020, 63, 203–216. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, L.; Huang, X.; Wang, X.; Yang, R.; Mao, J.; Wang, X.; Wang, X.; Zhang, Q.; Li, P. Identification of nutritional components in black sesame determined by widely targeted metabolomics and traditional Chinese medicines. Molecules 2018, 23, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dossou, S.S.K.; Xu, F.; You, J.; Zhou, R.; Li, D.; Wang, L. Widely targeted metabolome profiling of different colored sesame (Sesamum indicum L.) seeds provides new insight into their antioxidant activities. Food Res. Int. 2022, 151, 110850. [Google Scholar] [CrossRef] [PubMed]
- Dossou, S.S.K.; Xu, F.; Cui, X.; Sheng, C.; Zhou, R.; You, J.; Tozo, K.; Wang, L. Comparative metabolomics analysis of different sesame (Sesamum indicum L.) tissues reveals a tissue-specific accumulation of metabolites. BMC Plant Biol. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Brigante, F.I.; Mas, A.L.; Pigni, N.B.; Wunderlin, D.A.; Baroni, M.V. Targeted metabolomics to assess the authenticity of bakery products containing chia, sesame and flax seeds. Food Chem. 2020, 312, 126059. [Google Scholar] [CrossRef]
- Hemalatha, S.; Rao, M. Sesame lignans enhance antioxidant activity of vitamin E in lipid peroxidation systems. Mol. Cell. Biochem. 2004, 262, 195–202. [Google Scholar] [CrossRef]
- Kamal-Eldin, A.; Moazzami, A.; Washi, S. Sesame seed lignans: Potent physiological modulators and possible ingredients in functional foods nutraceuticals. Recent Pat. Food Nutr. Agric. 2011, 3, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.K.; Kar, J.; Sahu, D. Advances in Sesame (Sesamum indicum L.) Breeding. In Advances in Plant Breeding Strategies: Industrial and Food Crops; Springer: Cham, Switzerland, 2019; pp. 577–635. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivar | Zhongzhi 13 | Zhongzhi 13 | Swetha | Yuzhi 11 | Baizhima | Mishouzhima | Baizhima | Xiaozihei |
|---|---|---|---|---|---|---|---|---|
| Type | Modern cultivar | Modern cultivar, | Modern cultivar | Modern cultivar | Landraces | Landraces | ND | ND |
| Genome size (Mb) | 274 | 292 | 340 | 211 | 267 | 254 | 309 | 305 |
| Technology | Illumina | PacBio/Hi-C | Illumina | Illumina | Illumina | Illumina | PacBio/Hi-C | PacBio/Hi-C |
| Contig N50 (kb) | 52.2 | 1064.3 | 11.5 | 17.9 | 47.3 | 47.9 | 13,482 | 21,279 |
| Scaffold N50 (Mb) | 2.10 | 20.52 | 0.02 | 0.32 | - | - | 23.37 | 17.01 |
| GC content (%) | 35.22 | - | 35.00 | 35.10 | - | - | 35.44 | 35.93 |
| Coding gene | 27,148 | 28,406 | 41,859 | 26,022 | 31,558 | 30,995 | 24,345 | 25,265 |
| Average length per gene (bp) | 3171 | - | 4032 | 3623 | 3673 | 3700 | 3422 | 3112 |
| Reference | [4] | [5] | [6] | [6] | [6] | [6] | [7] | [8] |
| Gene Category | Genes |
|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Cytochrome | petA, * petB, * petD, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, *atpF, atpH, atpI |
| Rubisco | rbcL |
| NADH dehydrogenase | * ndhA,§,* ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Ribosomal protein (large subunit) | §,* rpl2, rpl14, * rpl16, rpl20, rpl22, § rpl23, rpl32, rpl33, rpl36 |
| Ribosomal protein (small subunit) | rps2, rps3, rps4, § rps7, rps8, rps11, §,*rps12, rps14, rps15, rps16, rps18, rps19 |
| RNA polymerase | rpoA, rpoB, * rpoC1, rpoC2 |
| ATP-dependent protease | * clpP |
| Cytochrome c biogenesis | ccsA |
| Membrane protein | cemA |
| Maturase | matK |
| Conserved reading frames | ycf1, § ycf2, ycf3, ycf4, § ycf15 |
| Translational initiation factor | infA |
| Pseudogenes | accD |
| Populations | Population Size | Number of Markers | Linkage Group Number | Total Length (cM) | QTL Manpping Reported | Reference |
|---|---|---|---|---|---|---|
| F2 population, COI1134 × RXBS | 96 lines | 8 EST-SSR, 25 AFLPs, 187 RSAMPLs | 30 | 936.72 | - | [24] |
| F2 population, COI1134 × RXBS | 260 lines | 30 EST-SSRs, 50 AFLPs, 573 RSAMPLs | 14 | 1216.00 | seed coat color | [25] |
| F6-RIL population, Zhongzhi 13 × Yiyangbai | 206 lines | 70 polymorphic SSRs, SRAPs and AFLPs | 15 | 592.4 | waterlogging tolerance | [26] |
| F8-RIL population, Zhongzhi 13 × ZZM2748 | 548 lines | 424 SSRs | 13 | 1869.80 | charcoal rot resistance; sesamin and sesamolin content | [27,28] |
| F2 population, Zhongzhi 13 × Shandong Jiaxiang Sesame | 107 lines | 1233 SLAFs | 15 | 1474.87 | - | [29] |
| F8-RIL population, Miaoqianzhima × Zhongzhi 14 | 224 lines | 1190 SNPs, 22 SSRs, 18 InDels | 14 | 844.46 | yield-related traits | [21] |
| F8-RIL poplation, Zhongzhi 13 × ZZM2748 | 430 lines | 1522 bins | 13 | 1090.99 | plant height, seed coat color | [34] |
| F6-RIL population, 95-223 × 92-3091 | 91 lines | 420 SNPs, 12 SSRs | 13 | 914 | - | [35] |
| BC1 population, Yuzhi 4 × BS | 300 lines | 9378 SLAFs | 13 | 1974.23 | basal branching habit, flowers per leaf axil | [36] |
| BC1 population, Yuzhi 4 × BS | 150 lines | 3528 SLAFs | 13 | 1312.52 | yield-related traits | [32] |
| F2 population, Gaoyou 8 × Ganzhi 6 | 122 lines | 2159 SNPs | 13 | 2128.51 | seed-related traits | [30] |
| F2 population, Muganli-57 × PI 599446 | 120 individuals | 782 SNPs | 13 | 697.3 | capsule shattering trait | [33] |
| F5-RIL population, Goenbaek × Osan | 90 lines | 1657 SNPs, 5 SSRs | 13 | 883.37 | phytophthora blight resistance | [31] |
| F2 population, Yuzhi DS899 × JS012 | 302 lines | 3041 bins (30,193 SNPs) | 13 | 2981.28 | determinacy trait | [37] |
| F2 population, cl1 × USA (0)-26 | 130 lines | 425,661 SNP/InDel variants | 13 | - | curly leaf and indehiscent capsule traits | [38] |
| F2 population, S-91 × S-297 | 149 lines | 2339 bins (3030 SNPs, 16,279 InDels) | 16 | 1497 | yield components, seed mineral-nutrients | [39] |
| F2 population, Yuzhi DS899 × JS012 | 120 individuals | 22,375 bins (380,544 SNP/InDel markers) | 13 | 1576.14 | seed coat color | [41] |
| F9-RIL population, Jinhuangma × Zhushanbai | 180 lines | 1354 bins (538,090 SNP/InDel variants) | 13 | 1295.45 | PEG-induced drought tolerance | [40] |
| Traits | Number of Accessions | Number of QTN | Number of Candidate Genes | Reference |
|---|---|---|---|---|
| The 56 agronomic traits: oil content, fatty acid biosynthesis and yield | 705 | 549 | 46 | [44] |
| drought/salt tolerance | 490 | 9/15 | 13/27 | [45] |
| seed yield-related | 705 | 547 | 48 | [47] |
| drought tolerance | 400 | 19 | 102 | [46] |
| tocopherol content | 96 | 1 | 1 | [51] |
| seven root traits | 327 | 19 | 32 | [48] |
| morpho-agronomic traits | 184 | 50 | 20 | [52] |
| phytophthora blight resistance | 87 | 29 | 34 | [31] |
| seed coat color | 366 | 224 | 92 | [53] |
| melatonin content | 450 | 3 | 14 | [49] |
| primary metabolite content | 412 | 433 | 10 | [50] |
| phytosterol contents | 402 | 33 | 37 | [54] |
| fatty acid composition and oil content | 400 | 43 | 20 | [55] |
| specific lignans | 410 | 89 | 10 | [56] |
| Stress | Varieties | Tissues | Descriptions | Reference |
|---|---|---|---|---|
| Salt stress | WZM3063 (ST), ZZM4028 (SS) | shoot of seedling | Transcriptome and metabolome profiles in the seedlings of salt-tolerant and sensitive sesame genotypes were performed in the early phase of salt stress. | [76] |
| WZM3063 (ST), ZZM4028 (SS) | shoot of seedling | miRNAs and their targets were identified from two contrasting sesame genotypes by a combined analysis of small RNAs and degradome sequencing. | [77] | |
| Drought stress | ZZM0635 (DT), ZZM4782 (DS) | root | Decipher the response of tolerant and sensitive genotypes to progressive drought and rewatering based on transcriptome. | [78] |
| ZZM3330 (DT), ZZM3743(DS) | leaf | Transcriptional and metabolic profiling in two sesame genotypes with contrasting ability to cope with drought stress. | [79] | |
| TEX-1 (DT), VEN-1 (DS) | root | Transcriptome analysis of two sesame genotypes with contrasting responses under PEG-induced osmotic stress. | [80] | |
| Waterlogging stress | Zhongzhi 13 (WT), ZZM0563 (WS) | root | RNA-seq-based analysis between waterlogging-tolerant and -susceptible genotypes. | [81] |
| ZZM2541 (WT), Ezhi3 (WS) | root | High-resolution temporal transcriptome analysis of two contrasting sesame genotypes over a 48 h period for waterlogging and drainage treatments. | [82] | |
| Heat stress | Taizhi3 (HT), SP19 (HS) | leaf | Transcriptome analysis of two sesame cultivars with different heat tolerance. | [83] |
| Disease stress | Yuzhi 11 (DT), RXBS (DS) | seedlings | Transcriptome profiles of resistant and susceptible sesame germplasm resources inoculated with Fusarium oxysporum f. sp. Sesami. | [84] |
| GT-10 (DT), RT-373 (DS) | root | Transcriptome analysis of resistant and susceptible sesame genotypes during Macrophomina phaseolina infection. | [85] |
| Traits | Varieties | Tissues | Number of DEGs | Number of Candidate Genes | Reference |
|---|---|---|---|---|---|
| Oil content | ZZM4728, ZZM3495, ZZM2161 | seeds, carpels | 794, 1807, 528 and 1667 of DEGs at 10, 20, 25,30 DPA | 23 sesame homologous lipid genes | [86] |
| Seed coat colors | Zhongfengzhi 1, Zhongzhi 33 | seed | the maximum DEGs at 11 DPA (20,253) | 20 genes | [87] |
| Seed coat colors | Yuzhi DS899, JS012 | seed | 2148, 5176, 3725, 2984 and 5115 of DEGs at 5, 10, 15, 20,25 DAF | 28 genes | [41] |
| Oil content and fatty acid composition | Zhongzhi 16, Mishuozhima | seed | 8404 DEGs | 20 genes | [55] |
| Leaf size | Zhongzhi 13, ZZM2289 | leaf | - | 26 genes | [88] |
| Root size | Baizhima, 697 | root | 1831 and 1066 up and down regulated genes | 10 genes | [89] |
| Male sterility | W1098A, W1098B | flower buds | 1502 DEGs | 49 homologous genes | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Tahir ul Qamar, M.; Yang, L.; Liang, J.; You, J.; Wang, L. Current Progress, Applications and Challenges of Multi-Omics Approaches in Sesame Genetic Improvement. Int. J. Mol. Sci. 2023, 24, 3105. https://doi.org/10.3390/ijms24043105
Li H, Tahir ul Qamar M, Yang L, Liang J, You J, Wang L. Current Progress, Applications and Challenges of Multi-Omics Approaches in Sesame Genetic Improvement. International Journal of Molecular Sciences. 2023; 24(4):3105. https://doi.org/10.3390/ijms24043105
Chicago/Turabian StyleLi, Huan, Muhammad Tahir ul Qamar, Li Yang, Junchao Liang, Jun You, and Linhai Wang. 2023. "Current Progress, Applications and Challenges of Multi-Omics Approaches in Sesame Genetic Improvement" International Journal of Molecular Sciences 24, no. 4: 3105. https://doi.org/10.3390/ijms24043105