
Complete Chloroplast Genome Sequences of Two Ehretia Trees (Ehretia cymosa and Ehretia obtusifolia): Genome Structures and Phylogenetic Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Samples and DNA Extraction
2.2. Sequencing and Assembly
2.3. Gene Annotation
2.4. Codon Usage and RNA Editing Sites
2.5. Repeat Analysis of Chloroplast Genomes
2.6. Characterization of Substitution Rate
2.7. Genome Comparison
2.8. Phylogenetic Analysis
3. Results
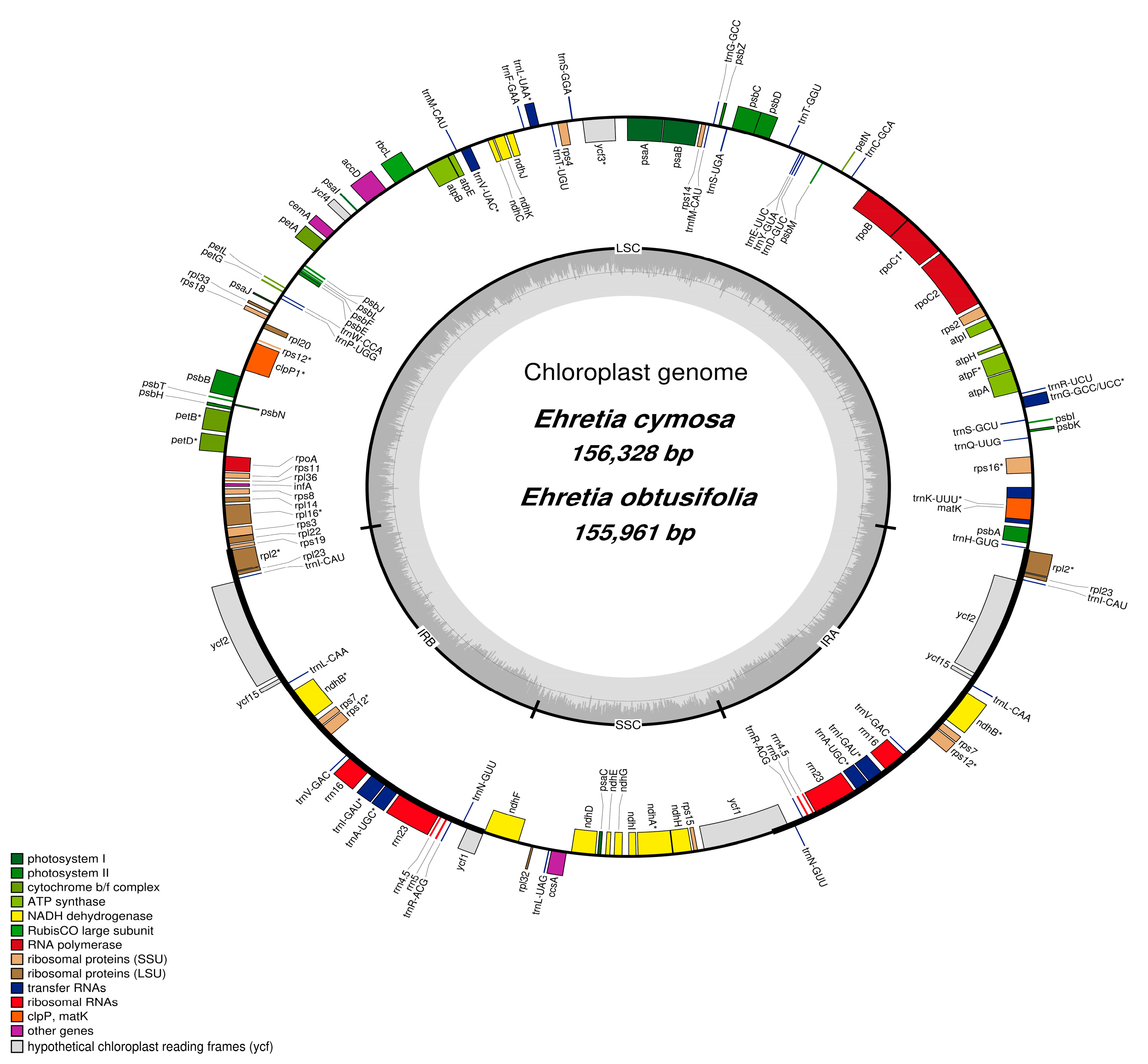
3.1. Characteristics of E. cymosa and E. obtusifolia
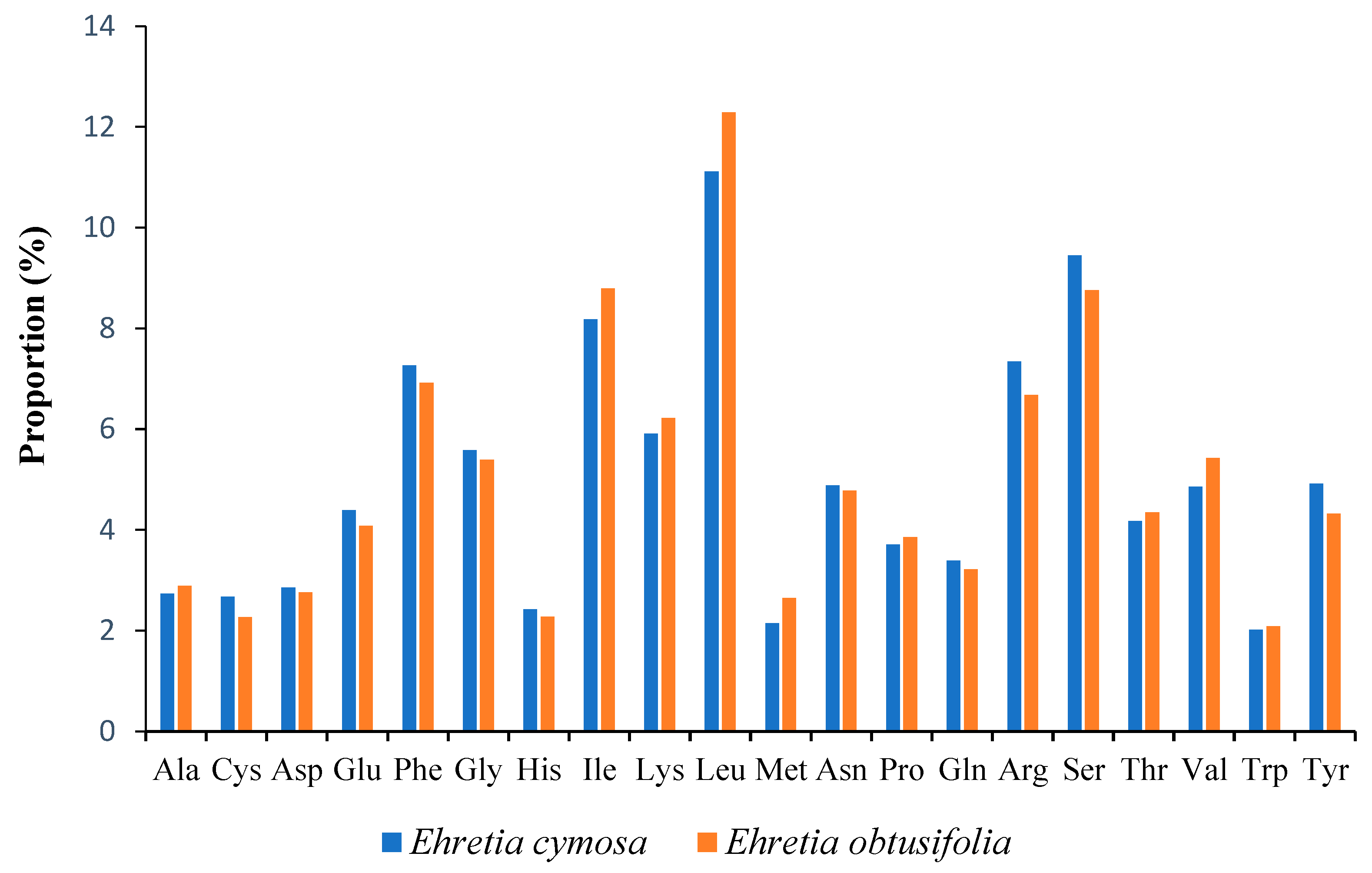
3.2. Codon Usage
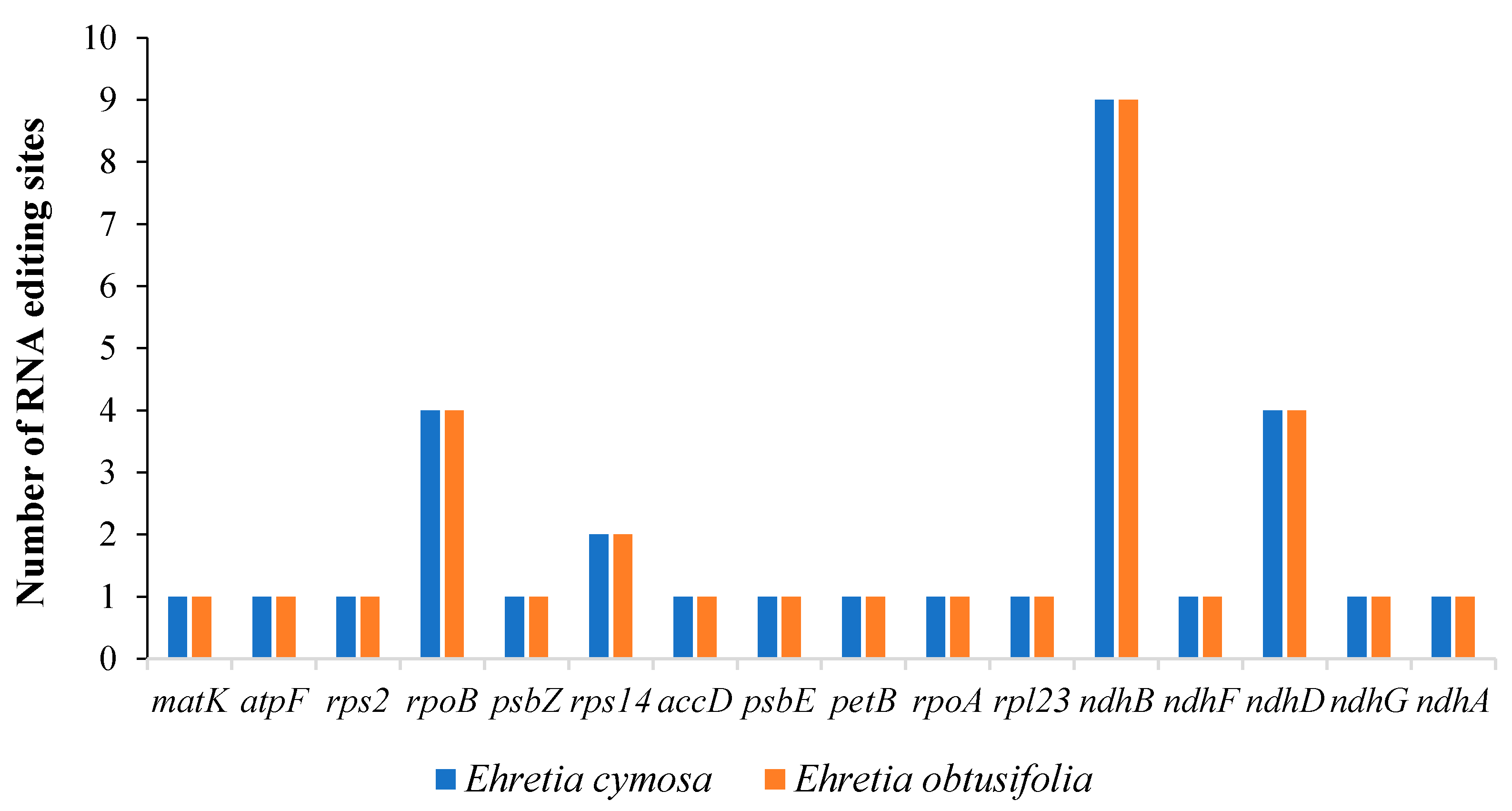
3.3. RNA Editing Sites
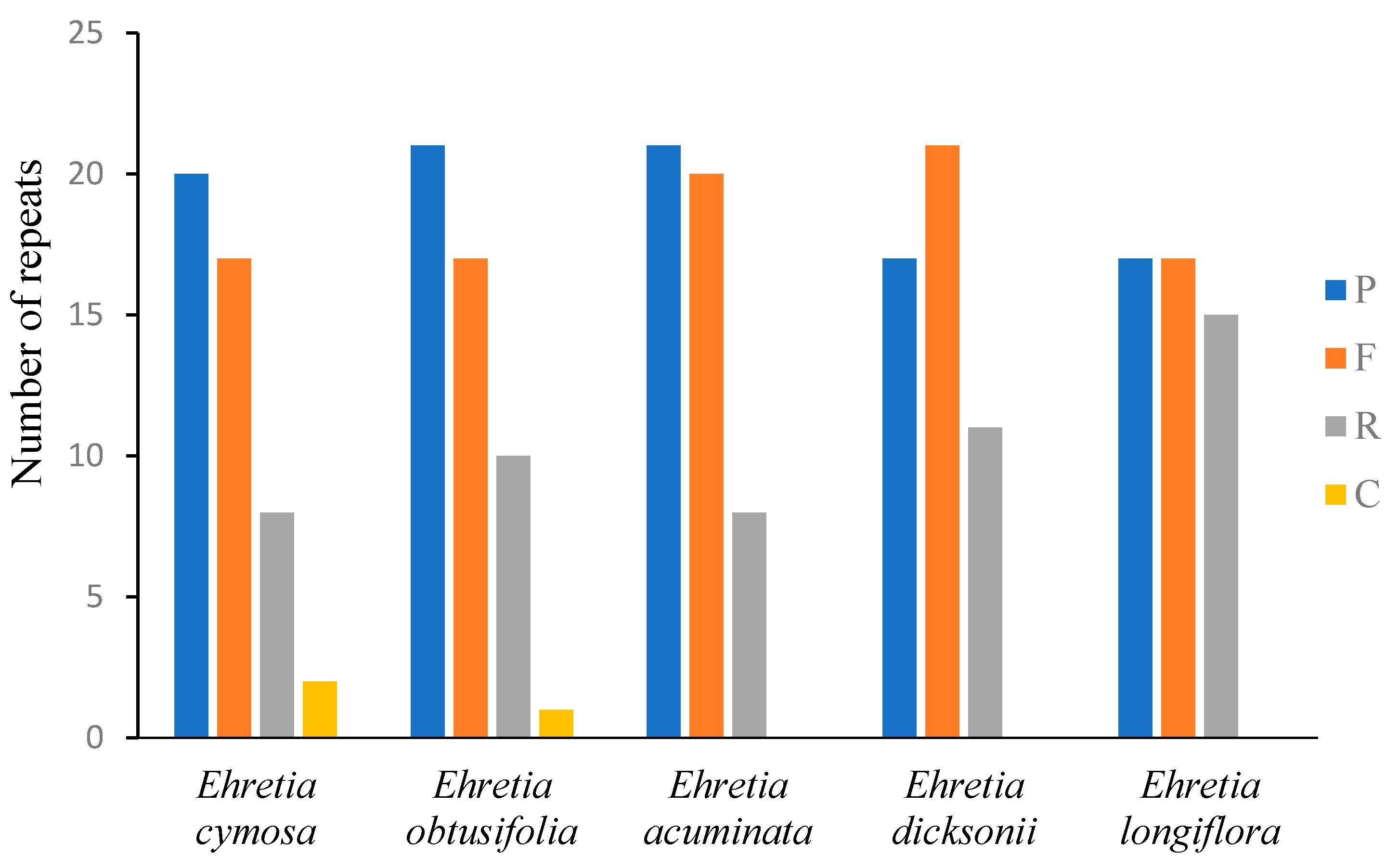
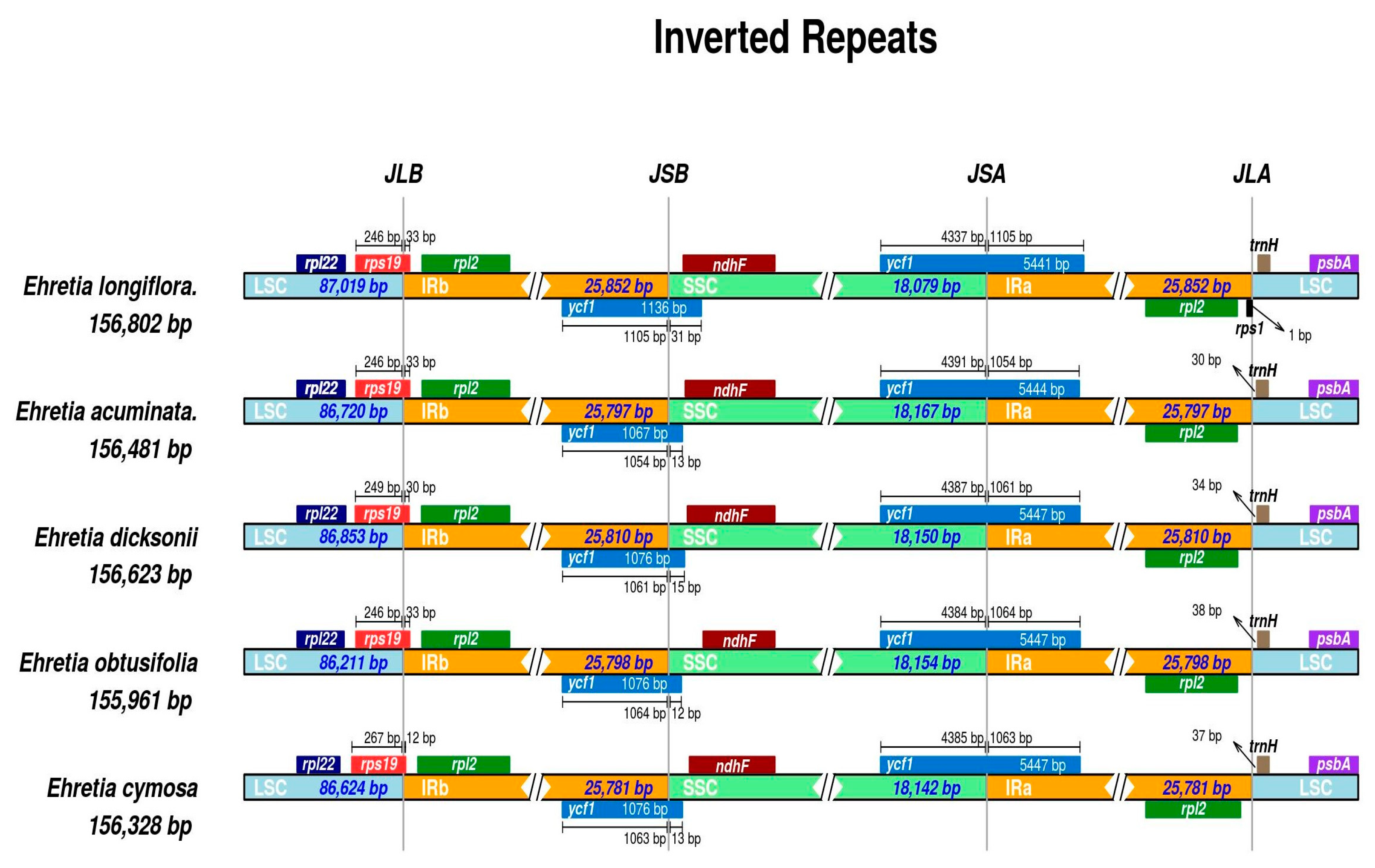
3.4. Long Repeats
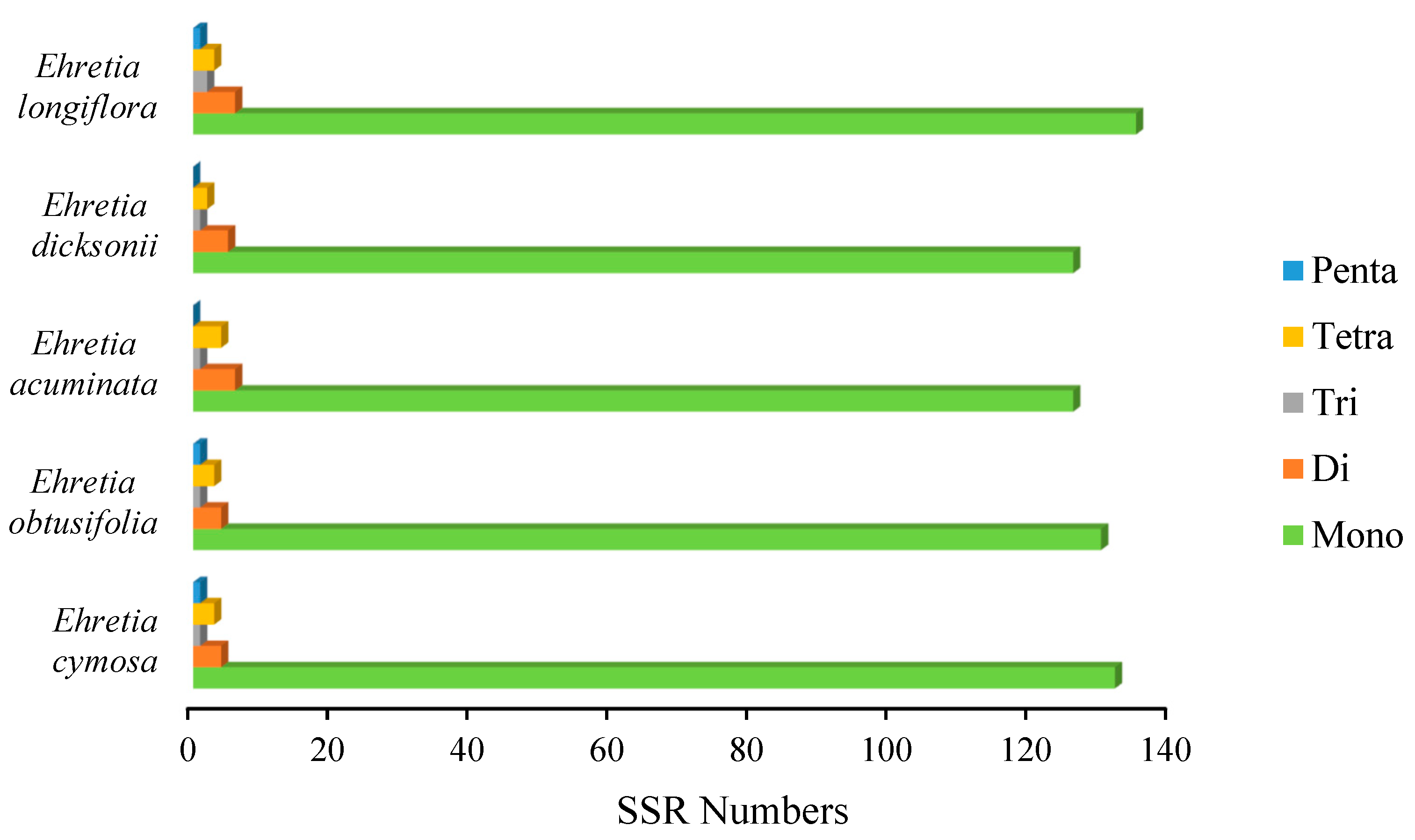
3.5. Simple Sequence Repeats
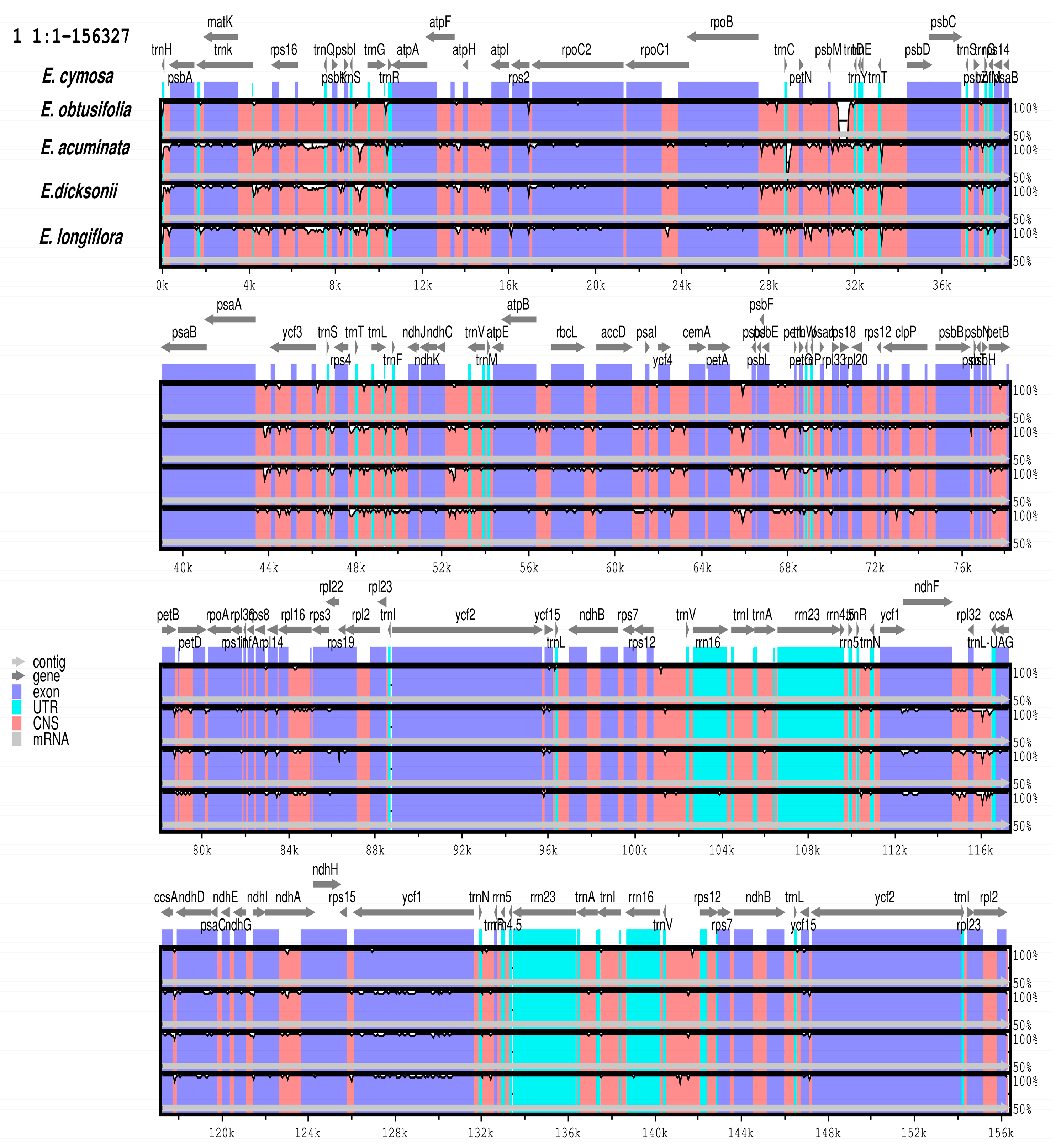
3.6. Comparative Analysis
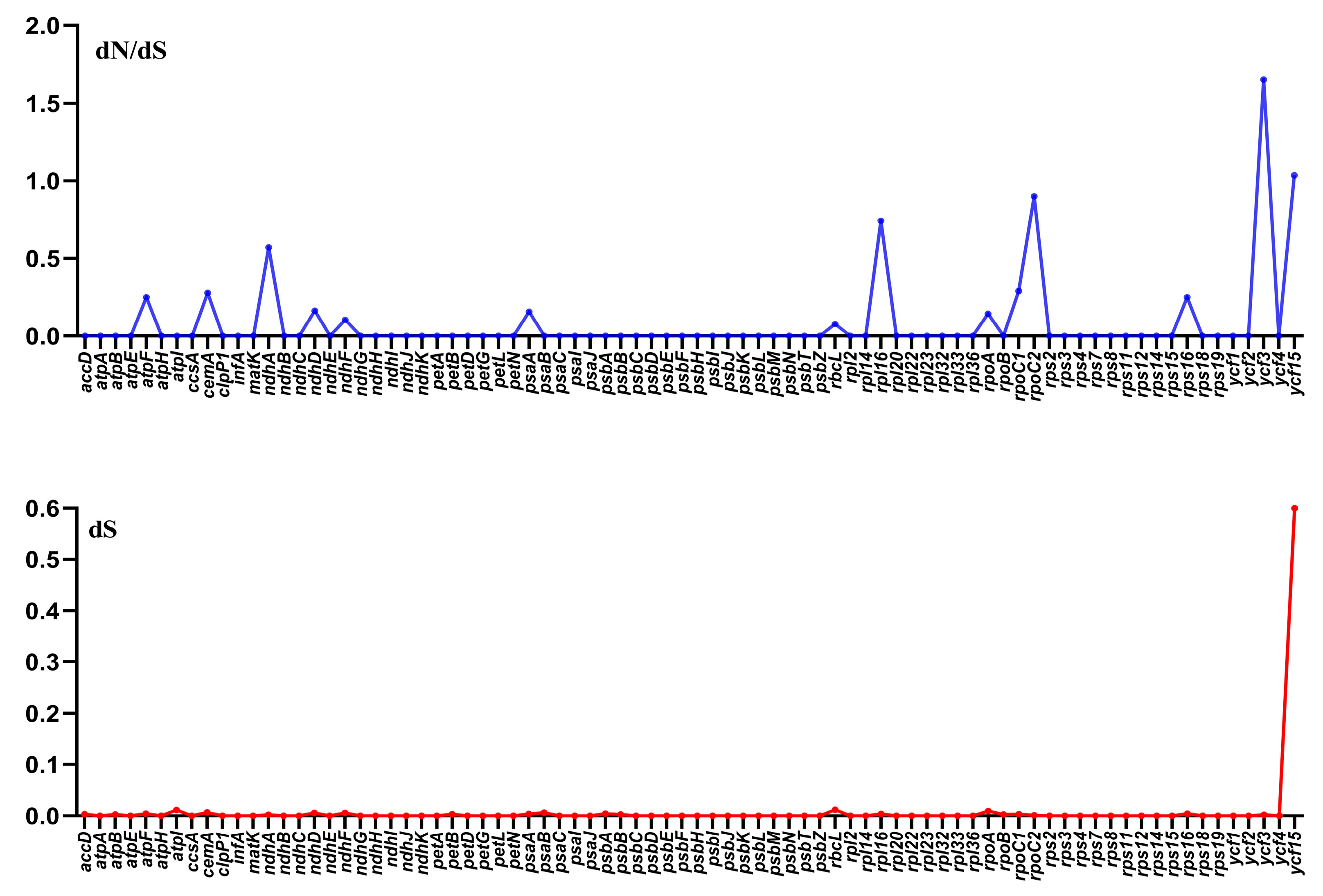
3.7. Divergence of Protein-Coding Gene Sequence
3.8. Characterization of the Substitution Rate
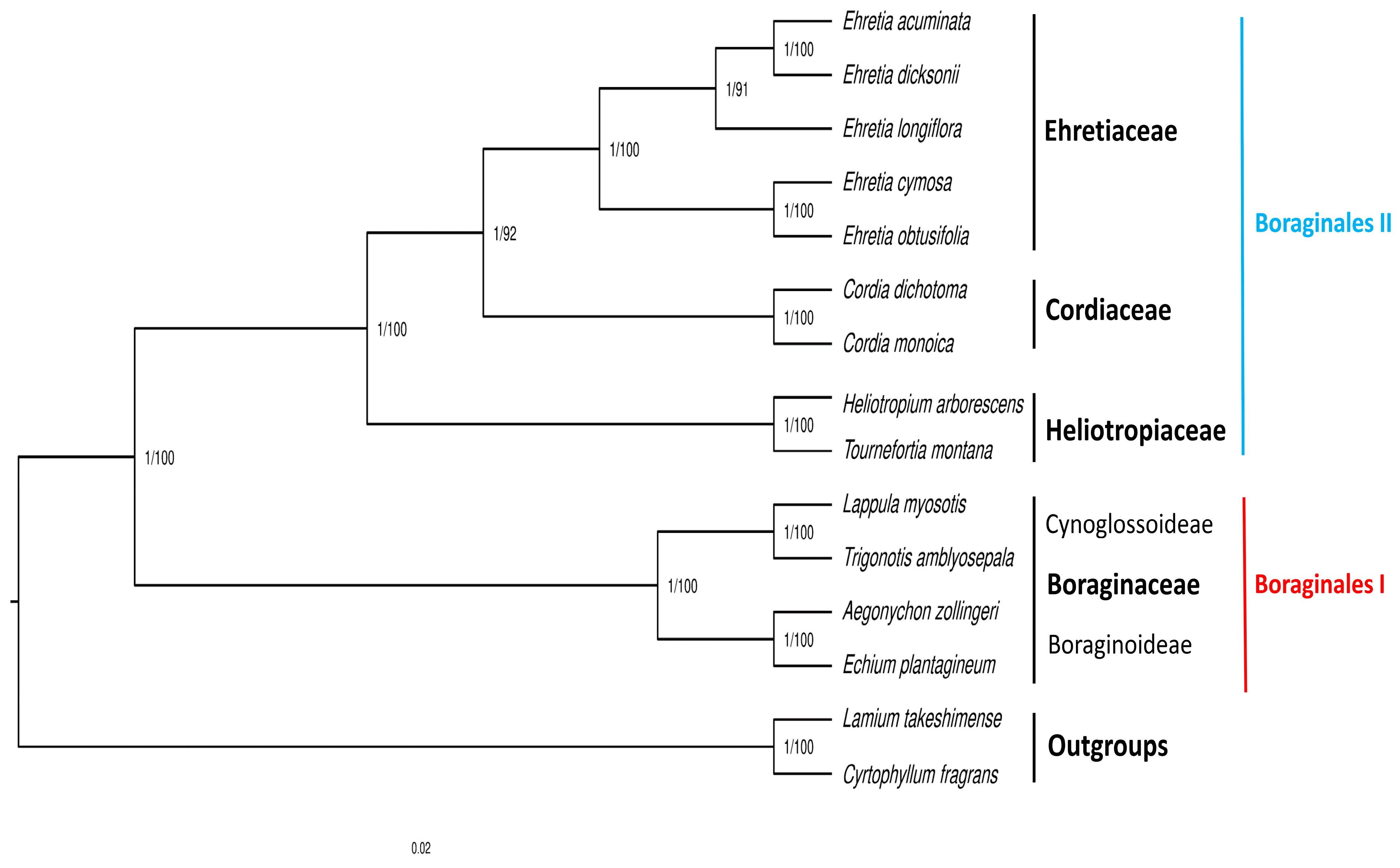
3.9. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Luebert, F.; Cecchi, L.; Frohlich, M.W.; Gottschling, M.; Guilliams, C.M.; Hasenstab-Lehman, K.E.; Hilger, H.H.; Miller, J.S.; Mittelbach, M.; Nazaire, M.; et al. Familial Classification of the Boraginales. Taxon 2016, 65, 502–522. [Google Scholar] [CrossRef] [Green Version]
- Heywood, V.H.; Brummitt, R.K.; Culham, A. Flowering Plant Families of the World; John Wiley: Hoboken, NJ, USA, 2007; ISBN 9781554072064. [Google Scholar]
- Simpson, M.G. Diversity and Classification of Flowering Plants: Eudicots. In Plant Systematics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 285–466. [Google Scholar]
- Candolle, A.P.d. Prodromus Systematis Naturalis Regni Vegetabilis, Sive, Enumeratio Contracta Ordinum Generum Specierumque Plantarum Huc Usque Cognitarium, Juxta Methodi Naturalis, Normas Digesta/Auctore Aug. Pyramo de Candolle. In Sumptibus Sociorum Treuttel et Würtz; De l’Ecole de Medecine: Paris, France, 1824. [Google Scholar]
- Engler, A.; Krause, K.; Pilger, R.; Prantl, K. Die Natürlichen Pflanzenfamilien Nebst Ihren Gattungen Und Wichtigeren Arten, Insbesondere Den Nutzpflanzen, Unter Mitwirkung Zahlreicher Hervorragender Fachgelehrten Begründet; Engelmann, W., Ed.; Verlag von Wilhelm Engelmann: Leipzig, Germany, 1887. [Google Scholar]
- Hutchinson, J. The Families of Flowering Plants. I. Dicotyledons. Arranged According to a New System Based on Their Probable Phylogeny. J. Hutchinson. Bot. Gaz. 1926, 82, 111–112. [Google Scholar] [CrossRef]
- Dahlgren, R.M.T. A Revised System of Classification of the Angiosperms. Bot. J. Linn. Soc. 1980, 80, 91–124. [Google Scholar] [CrossRef]
- Thorne, R. An Updated Phylogenetic Classification of the Flowering Plants. Aliso 1992, 13, 265–389. [Google Scholar] [CrossRef] [Green Version]
- Takhtajan, A. Diversity and Classification of Flowering Plants; Columbia University Press: New York, NY, USA, 1997; ISBN 9780231100984. [Google Scholar]
- The Angiosperm Phylogeny Group. An Ordinal Classification for the Families of Flowering Plants. Ann. Mo. Bot. Gard. 1998, 85, 531. [Google Scholar] [CrossRef] [Green Version]
- The Angiosperm Phylogeny Group. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG II. Bot. J. Linn. Soc. 2003, 141, 399–436. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Jansen, R.K. Molecular Evidence for the Age, Origin, and Evolutionary History of the American Desert Plant Genus Tiquilia (Boraginaceae). Mol. Phylogenetics Evol. 2006, 39, 668–687. [Google Scholar] [CrossRef] [PubMed]
- The Angiosperm Phylogeny Group. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Nazaire, M.; Hufford, L. A Broad Phylogenetic Analysis of Boraginaceae: Implications for the Relationships of Mertensia. Syst. Bot. 2012, 37, 758–783. [Google Scholar] [CrossRef]
- The Angiosperm Phylogeny Group. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gottschling, M.; Hilger, H.H.; Wolf, M.; Diane, N. Secondary Structure of the ITS1 Transcript and Its Application in a Reconstruction of the Phylogeny of Boraginales. Plant Biol. 2001, 3, 629–636. [Google Scholar] [CrossRef]
- Cohen, J.I. A Phylogenetic Analysis of Morphological and Molecular Characters of Boraginaceae: Evolutionary Relationships, Taxonomy, and Patterns of Character Evolution. Cladistics 2013, 30, 139–169. [Google Scholar] [CrossRef] [PubMed]
- Weigend, M.; Luebert, F.; Gottschling, M.; Couvreur, T.L.P.; Hilger, H.H.; Miller, J.S. From Capsules to Nutlets-Phylogenetic Relationships in the Boraginales. Cladistics 2013, 30, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Refulio-Rodriguez, N.F.; Olmstead, R.G. Phylogeny of Lamiidae. Am. J. Bot. 2014, 101, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Hasenstab-Lehman, K. Phylogenetics of the Borage Family: Delimiting Boraginales and Assessing Closest Relatives. Aliso 2017, 35, 41–49. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, T.; Luebert, F.; Xiang, Y.; Huang, C.-H.; Hu, Y.; Rees, M.; Frohlich, M.W.; Qi, J.; Weigend, M.; et al. Asterid Phylogenomics/Phylotranscriptomics Uncover Morphological Evolutionary Histories and Support Phylogenetic Placement for Numerous Whole-Genome Duplications. Mol. Biol. Evol. 2020, 37, 3188–3210. [Google Scholar] [CrossRef]
- Gottschling, M.; Luebert, F.; Hilger, H.H.; Miller, J.S. Molecular Delimitations in the Ehretiaceae (Boraginales). Mol. Phylogenetics Evol. 2014, 72, 1–6. [Google Scholar] [CrossRef]
- Grevich, J.J.; Daniell, H. Chloroplast Genetic Engineering: Recent Advances and Future Perspectives. CRC Crit. Rev. Plant Sci. 2005, 24, 83–107. [Google Scholar] [CrossRef]
- Roston, R.L.; Jouhet, J.; Yu, F.; Gao, H. Editorial: Structure and Function of Chloroplasts. Front Plant Sci. 2018, 9, 1656. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of Whole Chloroplast Genome Sequences to Choose Noncoding Regions for Phylogenetic Studies in Angiosperms: The Tortoise and the Hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P.; Vickrey, T.L. Chapter Nine-Structural Diversity Among Plastid Genomes of Land Plants. In Plastid Genome Evolution; Chaw, S.-M., Jansen, R.K.B.T.-A., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 85, pp. 263–292. ISBN 0065-2296. [Google Scholar]
- Bendich, A.J. Circular Chloroplast Chromosomes: The Grand Illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A Package for Visualizing Detailed Chloroplast Genome Structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhao, F.; Xu, Y.; Zhao, K.; Quan, H.; Su, Y.; Hao, P.; Liu, J.; Yu, B.; Yao, M.; et al. Complete Chloroplast Genome Sequencing and Phylogenetic Analysis of Two Dracocephalum Plants. Biomed. Res. Int. 2020, 2020, 4374801. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Chen, Y.Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo Assembly of Organelle Genomes from Whole Genome Data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Lenz, H.; Knoop, V. PREPACT 2.0: Predicting C-to-U and U-to-C RNA Editing in Organelle Genome Sequences with Multiple References and Curated RNA Editing Annotation. Bioinform. Biol. Insights 2013, 7, 1–19. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing Global DNA Sequence Alignments of Arbitrary Length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hou, B.-W.; Niu, Z.-T.; Liu, W.; Xue, Q.-Y.; Ding, X.-Y. Comparative Chloroplast Genomes of Photosynthetic Orchids: Insights into Evolution of the Orchidaceae and Development of Molecular Markers for Phylogenetic Applications. PLoS ONE 2014, 9, e99016. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.; Wang, Q.; Liu, C.; Zhang, R.; Cheng, A.; Sun, J. The Complete Chloroplast Genome Sequence of Borago Officinalis Linn. (Boraginaceae) and Its Phylogenetic Analysis. Mitochondrial DNA Part B 2020, 5, 1461–1462. [Google Scholar] [CrossRef] [Green Version]
- Carvalho Leonardo, I.; Barreto Crespo, M.T.; Capelo, J.; Bustos Gaspar, F. The Complete Plastome of Echium plantagineum L. (Boraginaceae), the First Chloroplast Genome Belonging to the Echium Genus. Mitochondrial DNA B Resour. 2022, 7, 1154–1156. [Google Scholar] [CrossRef]
- Wu, J.-H.; Li, H.-M.; Lei, J.-M.; Liang, Z.-R. The Complete Chloroplast Genome Sequence of Trigonotis Peduncularis (Boraginaceae). Mitochondrial DNA B Resour. 2022, 7, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wei, R. Comparison of Boraginales Plastomes: Insights into Codon Usage Bias, Adaptive Evolution, and Phylogenetic Relationships. Diversity 2022, 14, 1104. [Google Scholar] [CrossRef]
- Liu, K.; Wang, R.; Guo, X.-X.; Zhang, X.-J.; Qu, X.-J.; Fan, S.-J. Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes in Eragrostideae (Chloridoideae, Poaceae). Plants 2021, 10, 109. [Google Scholar] [CrossRef]
- Long, L.; Li, Y.; Wang, S.; Liu, Z.; Wang, J.; Yang, M. Complete Chloroplast Genomes and Comparative Analysis of Ligustrum Species. Sci. Rep. 2023, 13, 212. [Google Scholar] [CrossRef]
- Jansen, R.; Ruhlman, T. Genomics of Chloroplasts and Mitochondria; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Shaul, O. How Introns Enhance Gene Expression. Int. J. Biochem. Cell Biol. 2017, 91, 145–155. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.; Li, Y.; Qian, J.; Han, J. Chloroplast Genome of Aconitum Barbatum Var. Puberulum (Ranunculaceae) Derived from CCS Reads Using the PacBio RS Platform. Front. Plant Sci. 2015, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces That Influence the Evolution of Codon Bias. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Luo, C. Molecular and Functional Diversity of RNA Editing in Plant Mitochondria. Mol. Biotechnol. 2018, 60, 935–945. [Google Scholar] [CrossRef]
- Shikanai, T. RNA Editing in Plant Organelles: Machinery, Physiological Function and Evolution. Cell Mol. Life Sci. 2006, 63, 698–708. [Google Scholar] [CrossRef]
- Konhar, R.; Debnath, M.; Vishwakarma, S.; Bhattacharjee, A.; Sundar, D.; Tandon, P.; Dash, D.; Biswal, D. The Complete Chloroplast Genome of Dendrobium Nobile, an Endangered Medicinal Orchid from North-East India and Its Comparison with Related Dendrobium Species. PeerJ 2019, 7, e7756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme Reconfiguration of Plastid Genomes in the Angiosperm Family Geraniaceae: Rearrangements, Repeats, and Codon Usage. Mol. Biol. Evol. 2010, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yang, M.; Li, Y.; Jiang, M.; Liu, C.; He, M.; Wu, B. Chloroplast Genomes of Two Pueraria DC. Species: Sequencing, Comparative Analysis and Molecular Marker Development. FEBS Open Bio 2022, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Li, X.; Wu, Z.; Li, Z.; Hou, X.; Li, F.Y. Characterization and Comparative Analysis of Complete Chloroplast Genomes of Three Species from the Genus Astragalus (Leguminosae). Front. Genet. 2021, 12, 705482. [Google Scholar] [CrossRef]
- Gan, J.; Li, Y.; Tang, D.; Guo, B.; Li, D.; Cao, F.; Sun, C.; Yu, L.; Yan, Z. The Complete Chloroplast Genomes of Gynostemma Reveal the Phylogenetic Relationships of Species within the Genus. Genes 2023, 14, 929. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, D.-S.; Zou, L.; Yao, C.-Y. Comparison of Chloroplast Genomes and Phylogenomics in the Ficus Sarmentosa Complex (Moraceae). PLoS ONE 2022, 17, e0279849. [Google Scholar] [CrossRef]
- Contreras-Díaz, R.; Arias-Aburto, M.; van den Brink, L. Characterization of the Complete Chloroplast Genome of Zephyranthes Phycelloides (Amaryllidaceae, Tribe Hippeastreae) from Atacama Region of Chile. Saudi J. Biol. Sci. 2022, 29, 650–659. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast Microsatellites: New Tools for Studies in Plant Ecology and Evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Addisalem, A.B.; Esselink, G.D.; Bongers, F.; Smulders, M.J.M. Genomic Sequencing and Microsatellite Marker Development for Boswellia Papyrifera, an Economically Important but Threatened Tree Native to Dry Tropical Forests. AoB Plants 2015, 7, plu086. [Google Scholar] [CrossRef] [Green Version]
- Ebert, D.; Peakall, R. Chloroplast Simple Sequence Repeats (CpSSRs): Technical Resources and Recommendations for Expanding CpSSR Discovery and Applications to a Wide Array of Plant Species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Ishaq, M.N.; Ehirim, B.O.; Nwanyanwu, G.C.; Abubaka, R.I. DNA Fingerprinting Simple Sequence Repeat (SSR) Marker-Basedof Some Varieties of Rice (Oryza Sativa L.) Released in Nigeria. Afr. J. Biotechnol. 2019, 18, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete Chloroplast Genome Sequence of Magnolia Kwangsiensis (Magnoliaceae): Implication for DNA Barcoding and Population Genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative Chloroplast Genomics: Analyses Including New Sequences from the Angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Messing, J. High-Throughput Sequencing of Three Lemnoideae (Duckweeds) Chloroplast Genomes from Total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of All Ndh Genes as Determined by Sequencing the Entire Chloroplast Genome of the Black Pine Pinus Thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef]
- Yao, G.; Jin, J.-J.; Li, H.-T.; Yang, J.-B.; Mandala, V.S.; Croley, M.; Mostow, R.; Douglas, N.A.; Chase, M.W.; Christenhusz, M.J.M.; et al. Plastid Phylogenomic Insights into the Evolution of Caryophyllales. Mol. Phylogenet Evol. 2019, 134, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Sun, S.; Twyford, A.D.; Li, B.; Zhou, R.; Chen, S.; Gao, Q.; Favre, A. Lineage-specific Plastid Degradation in Subtribe Gentianinae (Gentianaceae). Ecol. Evol. 2021, 11, 3286–3299. [Google Scholar] [CrossRef]
- Xu, X.-M.; Liu, D.-H.; Zhu, S.-X.; Wang, Z.-L.; Wei, Z.; Liu, Q.-R. Phylogeny of Trigonotis in China—With a Special Reference to Its Nutlet Morphology and Plastid Genome. Plant Divers. 2023, in press. [Google Scholar] [CrossRef]
- Huo, Y.; Gao, L.; Liu, B.; Yang, Y.; Kong, S.; Sun, Y.; Yang, Y.; Wu, X. Complete Chloroplast Genome Sequences of Four Allium Species: Comparative and Phylogenetic Analyses. Sci. Rep. 2019, 9, 12250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhang, Y.; Xu, J.; Li, W.; Li, M. Characterization of the Complete Chloroplast Genome Sequence of Dalbergia Species and Its Phylogenetic Implications. Sci. Rep. 2019, 9, 20401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-F.; Landis, J.B.; Wang, H.-X.; Zhu, Z.-X.; Wang, H.-F. Comparative Analysis of Chloroplast Genome Structure and Molecular Dating in Myrtales. BMC Plant Biol. 2021, 21, 219. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the Most Promising Plastid DNA Barcode of Land Plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Chen, F.; Qin, P.; Xie, H.; Peng, G.; Li, Y.; Guo, X. The Specific DNA Barcodes Based on Chloroplast Genes for Species Identification of Theaceae Plants. Physiol. Mol. Biol. Plants 2022, 28, 837–848. [Google Scholar] [CrossRef]
- Shi, H.; Yang, M.; Mo, C.; Xie, W.; Liu, C.; Wu, B.; Ma, X. Complete Chloroplast Genomes of Two Siraitia Merrill Species: Comparative Analysis, Positive Selection and Novel Molecular Marker Development. PLoS ONE 2019, 14, e0226865. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Yang, J.; Sun, J.; Ju, M.; Zhao, Y.; Zhao, G. Comparative Analyses of Chloroplast Genomes of Cucurbitaceae Species: Lights into Selective Pressures and Phylogenetic Relationships. Molecules 2018, 23, 2165. [Google Scholar] [CrossRef] [Green Version]
- Chacón, J.; Luebert, F.; Hilger, H.H.; Ovchinnikova, S.; Selvi, F.; Cecchi, L.; Guilliams, C.M.; Hasenstab-Lehman, K.; Sutorý, K.; Simpson, M.G.; et al. The Borage Family (Boraginaceae s. Str.): A Revised Infrafamilial Classification Based on New Phylogenetic Evidence, with Emphasis on the Placement of Some Enigmatic Genera. Taxon 2016, 65, 523–546. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Li, S.; Wang, Y.; Wang, X. Comparative Genome/Transcriptome Analysis Probes Boraginales’ Phylogenetic Position, WGDs in Boraginales, and Key Enzyme Genes in the Alkannin/Shikonin Core Pathway. Mol. Ecol. Resour. 2019, 20, 228–241. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Ehretia cymosa | Ehretia obtusifolia |
|---|---|---|
| Genome size (bp) | 156,328 | 155,961 |
| IR (bp) | 25,781 | 25,798 |
| LSC (bp) | 86,624 | 86,211 |
| SSC (bp) | 18,142 | 18,154 |
| Total number of genes | 134 | 134 |
| rRNA | 4 | 4 |
| tRNA | 30 | 30 |
| Protein-coding genes | 80 | 80 |
| T (U) % | 31.40 | 31.40 |
| C % | 19.29 | 19.31 |
| A % | 30.72 | 30.70 |
| G % | 18.57 | 18.56 |
| Overall GC content % | 37.86 | 37.87 |
| GC content in LSC % | 35.91 | 35.91 |
| GC content in SSC % | 32.15 | 32.01 |
| GC content in IR % | 43.17 | 43.18 |
| SSR Type | Repeat Unit | Species | |
|---|---|---|---|
| E. cymosa | E. obtusifolia | ||
| Mono | A/T C/G | 130 2 | 128 2 |
| Di | AT/AT | 4 | 4 |
| Tri | AAG/CTT | 1 | 1 |
| Tetra | AAAC/GTTT AAAT/ATTT | 1 2 | 1 2 |
| Penta | AATCC/ATTGG | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alawfi, M.S.; Alzahrani, D.A.; Albokhari, E.J. Complete Chloroplast Genome Sequences of Two Ehretia Trees (Ehretia cymosa and Ehretia obtusifolia): Genome Structures and Phylogenetic Analysis. Forests 2023, 14, 1486. https://doi.org/10.3390/f14071486
Alawfi MS, Alzahrani DA, Albokhari EJ. Complete Chloroplast Genome Sequences of Two Ehretia Trees (Ehretia cymosa and Ehretia obtusifolia): Genome Structures and Phylogenetic Analysis. Forests. 2023; 14(7):1486. https://doi.org/10.3390/f14071486
Chicago/Turabian StyleAlawfi, Mohammad S., Dhafer A. Alzahrani, and Enas J. Albokhari. 2023. "Complete Chloroplast Genome Sequences of Two Ehretia Trees (Ehretia cymosa and Ehretia obtusifolia): Genome Structures and Phylogenetic Analysis" Forests 14, no. 7: 1486. https://doi.org/10.3390/f14071486