The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifolium alexandrinum and T. resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Features of the T. alexandrium and T. resupinatum cp Genomes
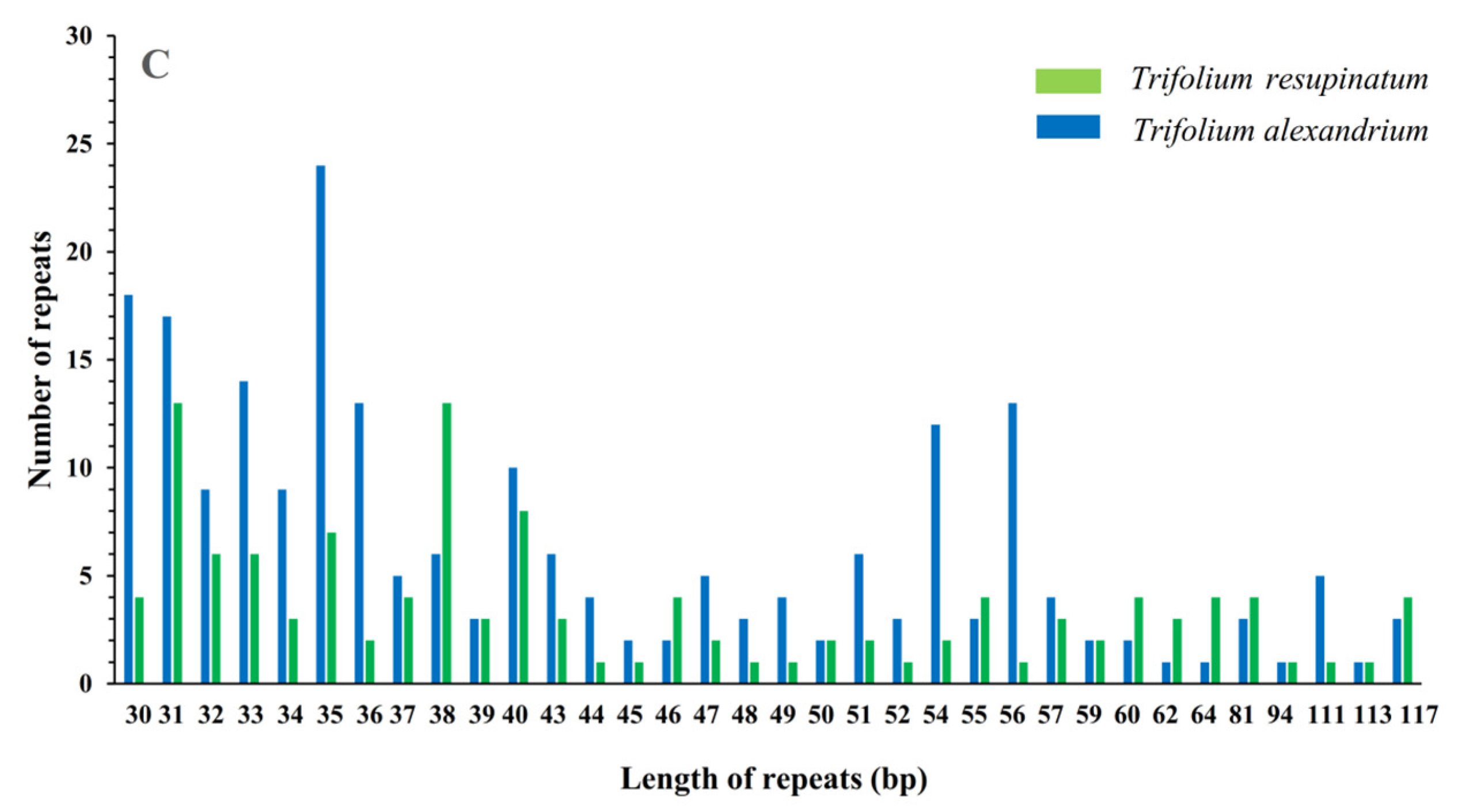
2.2. Repeat Sequences Analysis
2.3. Relative Synonymous Codon Usage Analysis
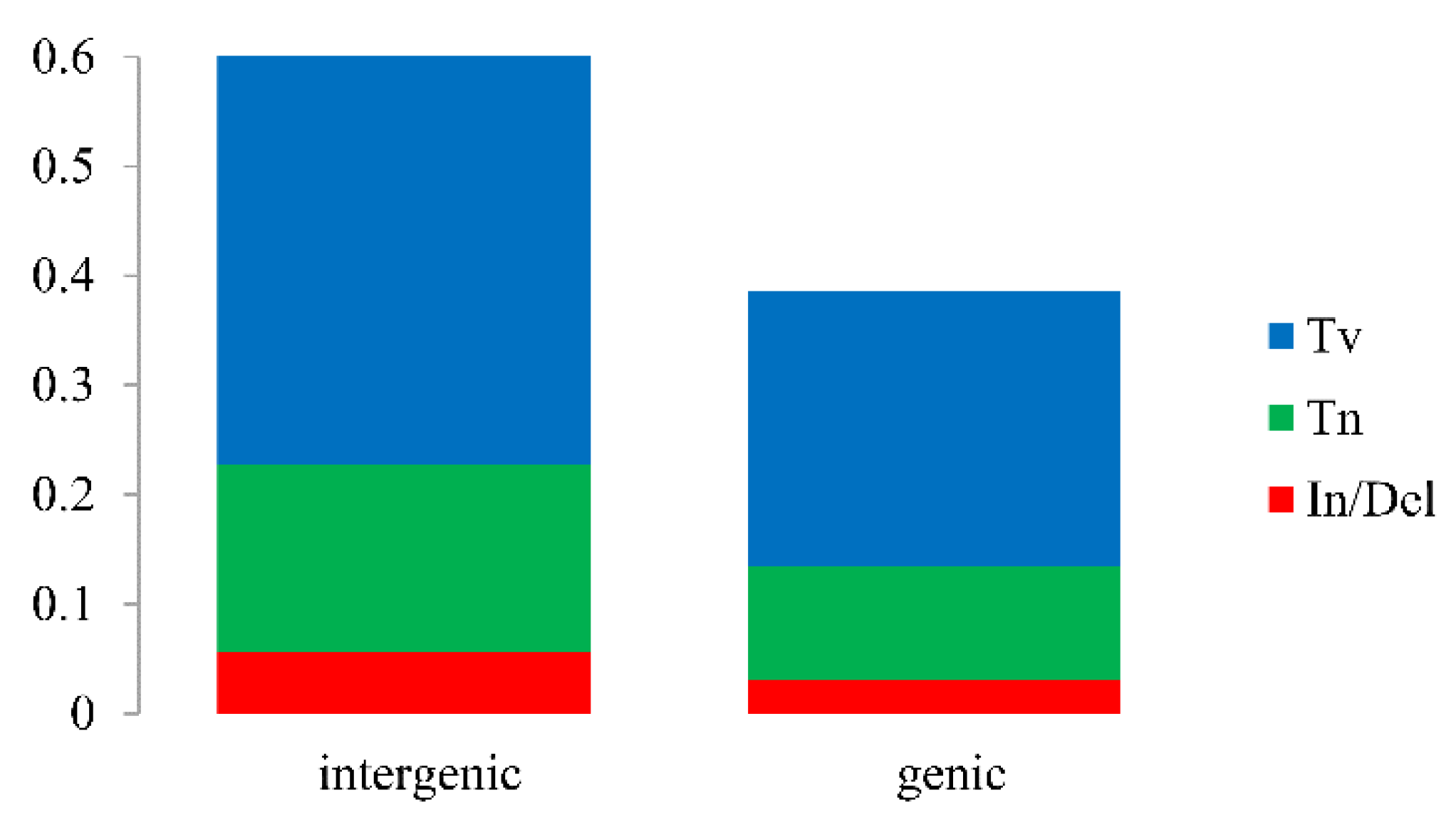
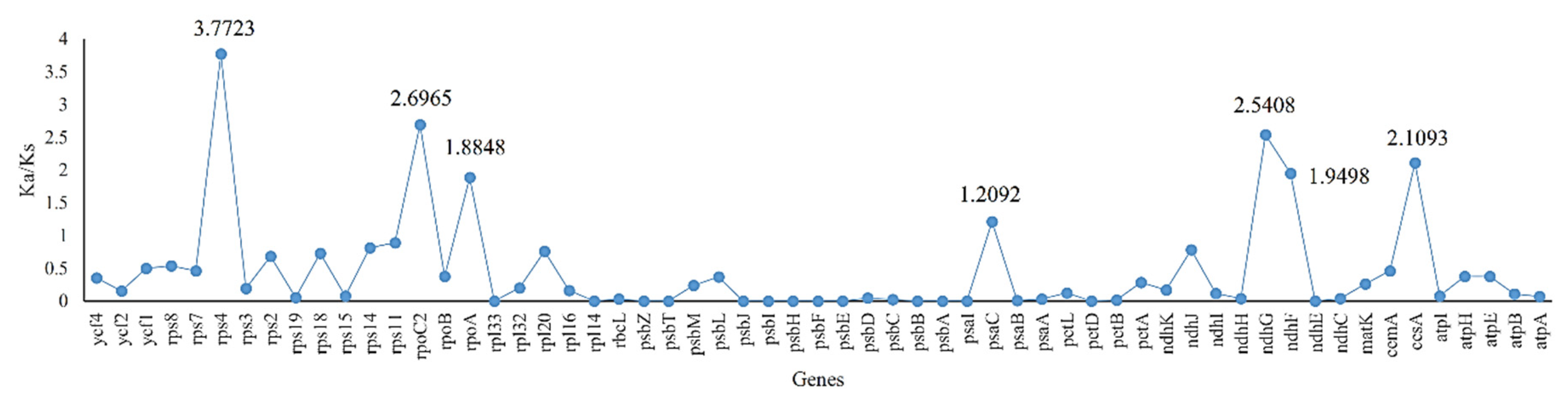
2.4. Ka/Ks, Single Nucleotide Polymorphisms (SNPs) and Insertions/Deletions (In/Dels)
2.5. Whole cp Genome Comparison with Other Trifolium Species
2.6. Phylogenetic Divergence Time Estimation
3. Discussion
3.1. Genome Feature of T. alexandrinum and T. resupinatum and Comparison with Other IR Lacking Trifolium Species
3.2. Relative Synonymous Codon Usage Analysis (RSCU)
3.3. SSRs and Large Repeat Sequences
3.4. Sequence Divergence and Hotspots
3.5. Phylogeny Analysis and Divergence Time
4. Methods
4.1. Plant Material, DNA Isolation and Sequencing
4.2. cp Genome Assembly, Annotation and Visualization
4.3. The Relative Synonymous Codon Usage Analysis (RSCU) and Simple Sequence Repeats (SSRs) Prediction
4.4. Sequence Variation Analysis and Ka/Ks
4.5. Divergence Time Estimates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
Abbreviations
| cp: | Chloroplast; |
| IRs | inverted-repeats; |
| IRLC: | IR lacking clade; |
| SNP: | single nucleotide polymorphisms; |
| In/Dels: | insertions/deletions; |
| ORF: | open reading frame; |
| SSC: | small single-copy region; |
| LSC: | large single-copy region; |
| NGS: | next generation sequencing; |
| Pi: | nucleotide diversity; |
| Ka: | non-synonymous; |
| Ks: | synonymous; |
| SSRs: | simple sequence repeats; |
| RSCU: | the relative synonymous codon usage analysis; |
| Tv: | transversion; |
| Tn: | transition; |
| CDS: | coding sequences; |
| CNS: | non-coding sequences; |
| UTR: | untranslated regions |
References
- Sabudak, T.; Guler, N. Trifolium L.—A review on its phytochemical and pharmacological profile. Phytother. Res. 2009, 23, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ellison, N.W.; Liston, A.; Steiner, J.J.; Williams, W.M.; Taylor, N.L. Molecular phylogenetics of the clover genus (Trifolium--Leguminosae). Mol. Phylogenet. Evol. 2006, 39, 688–705. [Google Scholar] [CrossRef] [PubMed]
- Badr, A.; El-Shazly, H.H.; Watson, L.E. Origin and ancestry of egyptian clover (Trifolium alexandrinum L.) as revealed by AFLP markers. Genet. Resour. Crop Evol. 2008, 55, 21–31. [Google Scholar] [CrossRef]
- Nazir, M.; Shah, F.H. Studies on persian clover (Trifolium resupinatum). Plant Food Hum. Nutr. 1985, 35, 57–62. [Google Scholar] [CrossRef]
- Douglas, S.E. Chloroplast origins and evolution. In The Molecular Biology of Cyanobacteria; Springer: Dordrecht, the Netherlands, 1994; pp. 91–118. [Google Scholar]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turmel, M.; Otis, C.; Lemieux, C. Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D.; Osorio, B.; Aldrich, J.; Thompson, W.F. Chloroplast DNA evolution among legumes: Loss of a large inverted repeat occurred prior to other sequence rearrangements. Curr. Genet. 1987, 11, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Turmel, M.; Otis, C.; Lemieux, C. Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, Ulvophyceae). Genome Biol. Evol. 2016, 8, 2789–2805. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and Cupressophytes and influence of heterotachy on the evaluation of Gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Kaila, T.; Chaduvla, P.K.; Rawal, H.C.; Saxena, S.; Tyagi, A.; Mithra, S.V.; Gaikwad, K. Chloroplast genome sequence of clusterbean (Cyamopsis tetragonoloba L.): Genome structure and comparative analysis. Genes 2017, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.F.; Freudenstein, J.V.; Li, J.; Mayfield-Jones, D.R.; Perez, L.; Pires, J.C.; Santos, C. Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol. Biol. Evol. 2014, 31, 3095–3112. [Google Scholar] [CrossRef] [Green Version]
- Sveinsson, S.; Cronk, Q. Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium). BMC Evol. Biol. 2014, 14, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberle, R.C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 2008, 66, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Gueutin, M.; Huang, X.; Higginson, E.; Ayliffe, M.; Day, A.; Timmis, J.N. Potential functional replacement of the plastidic acetyl-CoA carboxylase subunit (accD) gene by recent transfers to the nucleus in some angiosperm lineages. Plant Physiol. 2013, 161, 1918–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, V.; Mudd, E.A.; Lamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. Cell Mol. Biol. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Chuan, O.H.; Palmer, J.D. Mitochondrial gene transfer in pieces: Fission of the ribosomal protein gene rpl2 and partial or complete gene transfer to the nucleus. Mol. Biol. Evol. 2001, 18, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar] [CrossRef]
- Xi, L.; Zhang, T.C.; Qiao, Q.; Ren, Z.M.; Zhao, J.; Yonezawa, T.; Hasegawa, M.; Crabbe, M.J.; Li, J.Q.; Zhong, Y. Complete chloroplast genome sequence of holoparasite Cistanche deserticola (Orobanchaceae) reveals gene loss and horizontal gene transfer from its host Haloxylon ammodendron (Chenopodiaceae). PLoS ONE 2013, 8, e58747. [Google Scholar]
- Cai, Z.Q.; Guisinger, M.; Kim, H.G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef]
- Yang, Y.; Tao, Z.; Dong, D.; Jia, Y.; Li, F.; Zhao, G.F. Comparative analysis of the complete chloroplast Genomes of five Quercus species. Front. Plant Sci. 2016, 7, 944–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjad, A.; Muhammad, W.; Abdul, L.K.; Muhammad, A.K.; Sang-Mo, K.; Qari, M.I.; Raheem, S.; Saqib, B.; Byung-Wook, Y.; In-Jung, L. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 289–304. [Google Scholar]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L. Many parallel losses of infA from chloroplast DNA during Angiosperm evolution with multiple independent transfers to the nucleus. Cochrane Database Syst. Rev. 2001, 13, 645–658. [Google Scholar]
- Jansen, R.K.; Wojciechowski, M.F.; Sanniyasi, E.; Lee, S.B.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.L.; Ma, L.C.; Zhang, Z.S.; Liu, W.X.; Liu, Z.P. Characterization of the complete chloroplast genome of alfalfa (Medicago sativa) (Leguminosae). Gene Rep. 2017, 6, 67–73. [Google Scholar] [CrossRef]
- Shi, C.; Wang, S.; Xia, E.H.; Jiang, J.J.; Zeng, F.C.; Gao, L.Z. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 2016, 6, 30135. [Google Scholar] [CrossRef]
- Pasquale, L.C.; Domenico, D.P.; Donatella, D.; Giovanni, G.V.; Gabriella, S. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e120589. [Google Scholar]
- Xiao, L. Intra-genomic polymorphism in the internal transcribed spacer (ITS) regions of Cycas revoluta: Evidence of incomplete concerted evolution. Biodivers. Sci. 2009, 17, 476–481. [Google Scholar]
- Goremykin, V.V.; Hirsch-Ernst, K.I.; Wölfl, S.; Hellwig, F.H. Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal Angiosperm. Mol. Biol. Evol. 2003, 9, 1499–1505. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Li, Y.F.; Sylvester, S.P.; Li, M.; Zhang, C.; Li, X.; Duan, Y.F.; Wang, X.R. The complete plastid genome of agnolia zenii and genetic comparison to Magnoliaceae species. Molecules 2019, 24, 261. [Google Scholar] [CrossRef] [Green Version]
- Talat, F.; Wang, K. Comparative bioinformatics analysis of the chloroplast genomes of a wild diploid Gossypium and two cultivated allotetraploid species. Iran. J. Biotechnol. 2015, 13, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, G.L.; Dorman, H.E.; Buchanan, A.; Challagundla, L.; Wallace, L.E. A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2014, 2, 1400059. [Google Scholar] [CrossRef] [PubMed]
- Saski, C.; Lee, S.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.; Jansen, R.K. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Morgante, M.; Pfeiffer, A.; Costacurta, A.; Olivieri, A.M.; Rafalski, J.A. Polymorphic Simple Sequence Repeats in Nuclear and Chloroplast Genomes: Applications to the Population Genetics of Trees; Springer: Dordrecht, the Netherlands, 1996. [Google Scholar]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2017, 7, 1939–1955. [Google Scholar] [CrossRef] [Green Version]
- Ebert, D.; Peakall, R.O. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Tachida, H. Evolution of repeated sequences in non-coding regions of the genome. Jpn. J. Genet. 1993, 68, 549–565. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.L.; Wen, Z.F.; Liu, F.; Zhang, J.W.; Huang, W.G.; Lan, Y.P.; Cheng, L.L.; Cao, Q.C.; Hu, G.G.; Yun, C. Bioinformatics analysis of ycf1 gene in Corylus. J. Shanxi Agric. Sci. 2018, 46, 1244–1247. [Google Scholar]
- Raes, J.; Van, D.P.Y. Functional divergence of proteins through frameshift mutations. Trends Genet. 2005, 21, 428–431. [Google Scholar] [CrossRef]
- Bobrova, V.K.; Troitsky, A.V.; Ponomarev, A.G.; Antonov, A.S. Low-molecular-weight rRNAs sequences and plant phylogeny reconstruction: Nucleotide sequences of chloroplast 4.5S rRNAs from Acorus calamus (Araceae) and Ligularia calthifolia (Asteraceae). Plant Syst. Evol. 1987, 156, 13–27. [Google Scholar] [CrossRef]
- Masood, M.S.; Nishikawa, T.; Fukuoka, S.; Njenga, P.K.; Tsudzuki, T.; Kadowaki, K. The complete nucleotide sequence of wild rice (Oryza nivara) chloroplast genome: First genome wide comparative sequence analysis of wild and cultivated rice. Gene 2004, 340, 133–139. [Google Scholar] [CrossRef]
- Su, H.J.; Hogenhout, S.A.; Al-Sadi, A.M.; Kuo, C.H. Complete chloroplast genome sequence of Omani lime (Citrus aurantiifolia) and comparative analysis within the Rosids. PLoS ONE 2014, 11, e113049. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Dong, W.P.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Richard, T.C. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662–670. [Google Scholar] [CrossRef] [Green Version]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin-A-Woeng, T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Yi, L. Comparing and analyzing on models of calculation and statistical testing of nonsynonymous substitution rate and synonymous substitution rate during gene evolution. J. Qujing Norm. Univ. 2006, 25, 1–6. [Google Scholar]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef]
- Bittner-Eddy, P.D.; Crute, I.R.; Holub, E.B.; Beynon, J.L. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica. Plant J. 2010, 21, 177–188. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of Orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. Collection and Annotation of Suinong14 Full-Length Transcripts and Gene Diversity Analysis of Glyma13g21630; Chinese Academy of Agricultural Science: Beijing, China, 2011.
- Malaviya, D.R.; Roy, A.K.; Kaushal, P.; Kumar, B.; Tiwari, A. Genetic similarity among Trifolium species based on isozyme banding pattern. Plant Syst. Evol. 2008, 276, 125–136. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Pyshkin, A.V. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Acemel, R.D.; Tena, J.J.; Irastorza-Azcarate, I.; Marlétaz, F.; Gómez-Marín, C.; Calle-Mustienes, E.; Mangenot, S. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet. 2016, 48, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13, S8. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Thierer, T. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; McVean, G. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genome Length(bp) | GC Content (%) | Gene Density | tRNA | rRNA | mRNA | Genes | Genes with Introns | GenBank Number | |
|---|---|---|---|---|---|---|---|---|---|---|
| Repetitive % | cp Genome | CDS | ||||||||
| T. alexandrinum | 148545/2.85% | 34.09 | 37.05 | 7.54 × 10−4 | 31 | 6 | 75 | 112 | 13 | MN857160 |
| T. resupinatum | 149026/2.69% | 33.80 | 36.64 | 7.31 × 10−4 | 37 | 6 | 66 | 109 | 5 | MN857161 |
| T. subterraneum | 144763/20.71% | 34.83 | 37.10 | 7.60 × 10−4 | 30 | 4 | 76 | 110 | 16 | NC011828 |
| T. meduseum | 142595/12.83% | 34.87 | 37.34 | 7.78 × 10−4 | 30 | 4 | 77 | 111 | 15 | NC476730.1 |
| T. pratense | 121178/NA * | 34.63 | 36.94 | 7.43 × 10−4 | 28 | 4 | 58 | 90 | 11 | KJ788290 |
| T. repens | 132120/20.70% | 34.53 | 36.96 | 8.10 × 10−4 | 31 | 4 | 72 | 107 | 16 | KC894706.1 |
| T.strictum | 125834/0.71% | 34.98 | 36.70 | 8.82 × 10−4 | 31 | 5 | 75 | 111 | 18 | NC025745.1 |
| T.aureum | 126970/5.60% | 34.86 | 36.81 | 8.51 × 10−4 | 30 | 4 | 74 | 108 | 15 | KC894708.1 |
| T.boissieri | 125740/1.05% | 35.24 | 36.83 | 8.75 × 10−4 | 31 | 5 | 74 | 110 | 17 | NC025743.1 |
| T.glanduliferum | 126149/0.78% | 34.90 | 36.70 | 8.72 × 10−4 | 30 | 5 | 75 | 110 | 17 | NC025744.1 |
| T. grandiflorum | 126149/5.60% | 37.20 | 35.82 | 8.80 × 10−4 | 30 | 4 | 77 | 111 | 16 | NC_024034 |
| T. hybridum | 134831/7.86% | 34.33 | 35.97 | 8.08 × 10−4 | 31 | 4 | 74 | 109 | 17 | KJ788286 |
| T. lupinaster | 135049/5.98% | 33.97 | 35.71 | 8.15 × 10−4 | 30 | 4 | 76 | 110 | 15 | KJ788287 |
| T. occidentale | 133780/4.64% | 36.34 | 35.91 | 8.00 × 10−4 | 29 | 4 | 74 | 107 | 17 | KJ788289 |
| T. semipilosum | 138194/10.55% | 36.31 | 35.81 | 7.89 × 10−4 | 31 | 4 | 74 | 109 | 17 | KJ788291 |
| Category | Function | Name of Genes | |||||
|---|---|---|---|---|---|---|---|
| Self-replication (31) | Ribosomal RNA Genes | rrn4.5 | rrn5 | rrn16 | rrn23 | ||
| Transfer RNA genes | trnA-ACG | trnA-GUC | trnA-GUU | trnA-UCU | trnA-UGC* | trnC-GCA | |
| trnG-GCC | trnG-UUC* | trnG-UUG | trnH-GUG | trnL-CAA | trnL-UAA* | ||
| trnL-UAG | trnL-UUU* | trnM-CAU | trnP-GAA | trnP-UGG | trnS-GCU | ||
| trnS-GGA | trnS-UGA | trnT-CCA | trnT-CGU* (ale) | trnT-GGU | trnT-GUA | ||
| trnT-UGU | trnV-GAC | trnV-UAC* | |||||
| Ribosomal proteins (10) | Small subunit of ribosome (SSU) | rps2 | rps3 | rps4 | rps7 | rps8 | rps11 |
| rps14 | rps15 | rps18*/ale | rps19 | ||||
| Transcription (12) | Large subunit of ribosome (LSU) | rpl2 (ale) | rpl14 | rpl16 | rpl20 | rpl23 | rpl32 |
| rpl33 | rpl36 | ||||||
| RNA polymerase subunits | rpoA | rpoB | rpoC1* (ale) | rpoC2 | |||
| Photosynthesis related genes (46) | Large subunit of Rubisco | rbcL | |||||
| Subunits of Photosystem I | psaA | psaB | psaC | psaI | psaJ | ||
| Subunits of Photosystem II | psbA | psbB | psbC | psbD | psbE | psbF | |
| psbH | psbI | psbJ | psbK | psbL | psbM | ||
| psbN (ale) | psbT | psbZ | |||||
| Subunits of ATP synthase | atpA | atpB | atpE | atpF* (ale) | atpH | atpI | |
| Cytochrome b/f complex | petA | petB | petD | petG | petL | petN | |
| C-type cytochrome synthesis gene | ccsA | ||||||
| Subunits of NADH dehydrogenase | ndhA* (ale) | ndhB* (ale) | ndhC | ndhD | ndhE | ndhF | |
| ndhG | ndhH | ndhI | ndhJ | ndhK | |||
| Other genes (7) | Maturase | matK | |||||
| Protease | clpP* (ale) | ||||||
| Chloroplast envelope membrane protein | cemA | ||||||
| Subunit of acetyl-CoA | accD (ale) | ||||||
| Hypothetical open reading frames | ycf1 | ycf2 | ycf3** (ale) | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; Xiong, Y.; He, J.; Yu, Q.; Zhao, J.; Lei, X.; Dong, Z.; Yang, J.; Peng, Y.; Zhang, X.; et al. The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifolium alexandrinum and T. resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae. Plants 2020, 9, 478. https://doi.org/10.3390/plants9040478
Xiong Y, Xiong Y, He J, Yu Q, Zhao J, Lei X, Dong Z, Yang J, Peng Y, Zhang X, et al. The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifolium alexandrinum and T. resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae. Plants. 2020; 9(4):478. https://doi.org/10.3390/plants9040478
Chicago/Turabian StyleXiong, Yanli, Yi Xiong, Jun He, Qingqing Yu, Junming Zhao, Xiong Lei, Zhixiao Dong, Jian Yang, Yan Peng, Xinquan Zhang, and et al. 2020. "The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifolium alexandrinum and T. resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae" Plants 9, no. 4: 478. https://doi.org/10.3390/plants9040478