![[全合成1]天然产物(±)-Steenkrotin A 的全合成(一)](data:image/svg+xml;utf8,<svg xmlns='http://www.w3.org/2000/svg'></svg>)
[全合成1]天然产物(±)-Steenkrotin A 的全合成(一)

天然产物(±)-Steenkrotin A是2008年Hussein课题组从大戟科巴豆属植物Croton steenkampianus树叶中分离得到的一种新型二萜化合物,含有复杂的高张力[3,5,5,6,7]五环骨架和8个手性中心。(其中六个连续)。




2015年浙江大学丁寒锋教授课题组从简单原料出发以18步反应高效完成(±)-Steenkrotin A的首次全合成。
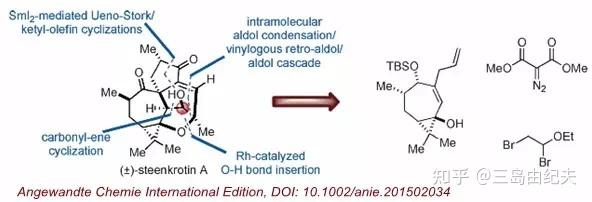

详细路线如下:
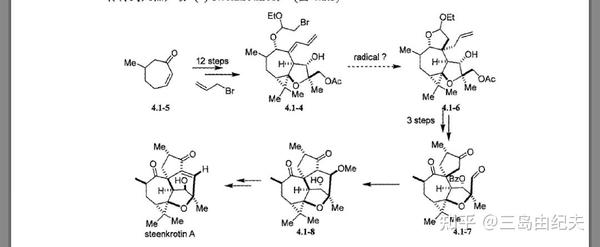

首先构建[3,5,7]三环骨架。
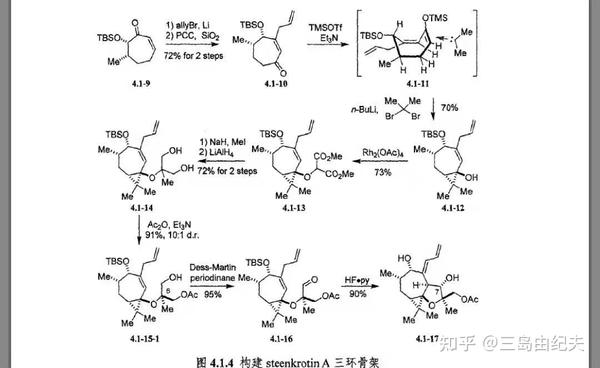

先从底物4.1-9 2-环庚烯-1-酮出发,烯丙基溴与金属锂原位生成烯丙基锂后与α,β-不饱和酮发生1,2-加成生成烯丙基叔醇。对于α,β-不饱和酮,有机锂试剂主要生成1,2加成产物,而格式试剂多生成1,2与1,4加成混合产物,而有机铜试剂主要生成1,4-加成产物,有机铜试剂可由有机锂试剂或格式试剂与Cu(Ι)盐原位反应生成得到。这一现象可由软硬酸碱理论解释,有机锂是“硬”亲核试剂更偏向于与“硬”的亲核位点羰基结合,有机铜试剂(比如二烷基铜锂)是更“软”的亲核试剂偏向于与更“软”的亲核位点共轭双键结合。产物烯丙基叔醇后在PCC/SiO2作用下发生Dauben-Michno氧化重排生成4.1-10。
Dauben-Michno氧化重排是PCC氧化应用的扩展。底物烯丙基叔醇经过PCC氧化重排发生1,3-羰基转移。反应机理是首先烯丙基叔醇在酸催化下生成烯丙基碳正离子,后羟基从位阻小的C3位进攻生成烯丙基仲醇并伴随着原双键的迁移,最后PCC氧化烯丙基仲醇,生成新的α,β-不饱和酮。PCC是强酸性氧化剂,有可能实现这三步的一步进行。
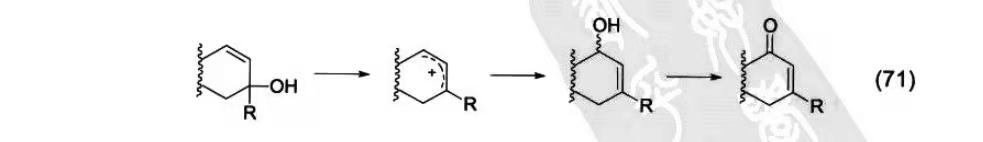

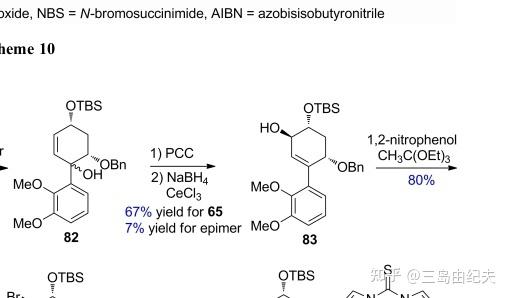
Dauben-Michno氧化重排在复杂天然产物全合成中得到了广泛的应用。比如在(-)-Galanthamine全合成中,通过Dauben-Michno氧化重排与Luche还原两步反应得到关键反应前体手性烯丙基醇83。

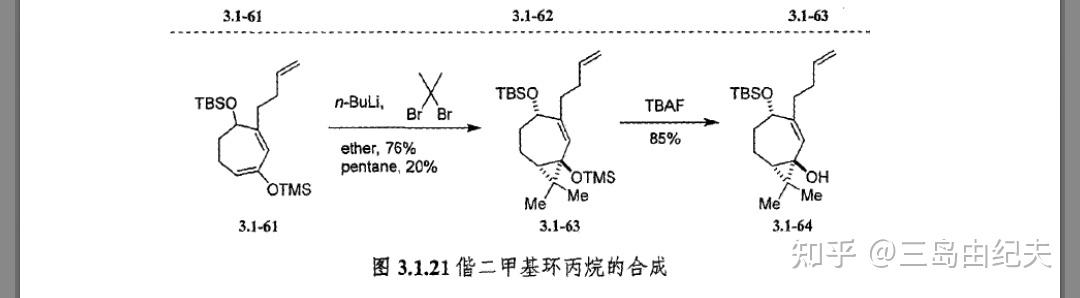
上述两步以总共72%产率得到4.1.10。然后后构建三元环,这里使用了经典也很常用的策略:卡宾或氮烯与烯烃的[2+1]环加成反应合成环丙烷或氮杂环丙烷。4.1-10先是在有机碱Et3N条件下与TMSOTf生成烯醇硅醚4.1-11,后正丁基锂与2,2-二溴丙烷发生α-脱卤消去生成二甲基卡宾后与烯醇硅醚发生[2+1]环加成,这一步中4.1-11七元环以类似船式的优势构象参与反应,因为4.1-11 C4位-OTBS较大,空间位阻较大,处于e键才更为稳定,后用HCl或TBAF处理脱去TMS保护基(这一步在图4.1.4中并没有表示),得到[3,7]二环叔醇4.1-12。三步产率70%。

4.1-12与重氮丙二酸二甲酯在Rh2(OAc)4(二聚醋酸铑)催化下发生铑卡宾的分子间的O-H键插入,以73%的产率得到产物二叔基醚4.1-13。以重氮化合物为金属卡宾前体对X-H(X=C,O,N,S,Si等)发生的插入反应在全合成中应用广泛。 常用于形成金属卡宾做催化剂的金属盐是二价铑盐或二价铜盐。
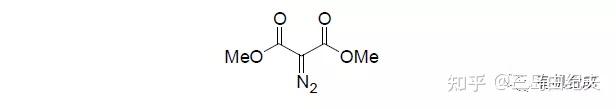

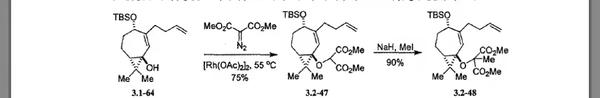

以α-重氮羰基化合物(比如重氮丙二酸二甲酯)为前体的铑卡宾对于羟基O-H键的插入是合成醚的重要方法。
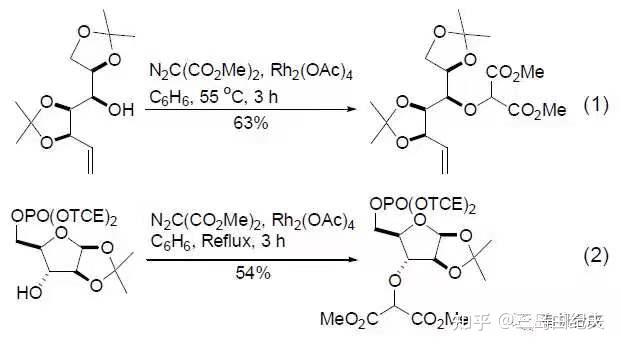

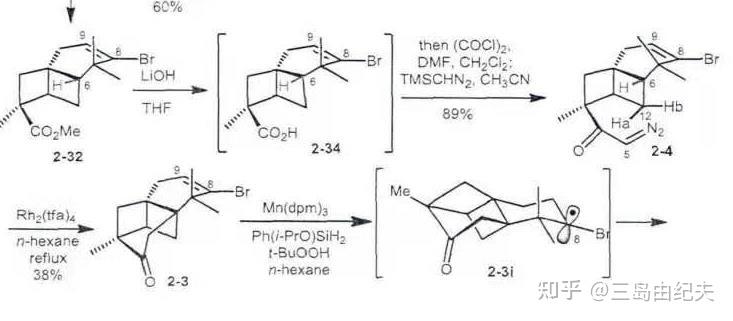
铑卡宾分子内对于C-H键的插入具有高效和较高立体选择性的优点,在全合成中应用广泛。一般进行1,5-插入或者1,6-插入,生成六元环或五元环。插入反应活性3º>2º>1º 。比如在天然产物(±)-Aplydactone全合成中关键步骤就是以α-重氮酮为铑卡宾前体对刚性骨架叔碳的C-H键的1,6-插入,构建了六元桥环骨架。

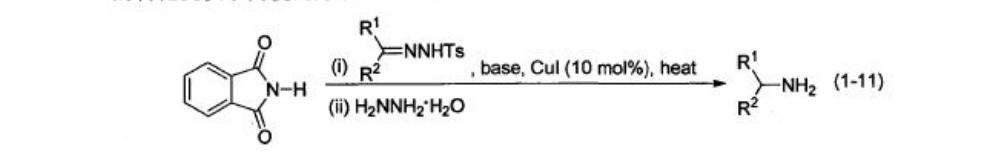
金属卡宾对N-H键的插入。比如N-对甲苯磺酰腙在强碱条件下分解原位生成重氮化合物,与CuI产生铜卡宾,与邻苯二甲酰亚胺发生N-H插入反应,再发生肼解生成1-11,为由醛酮转化为胺提供了一种可行的方法。

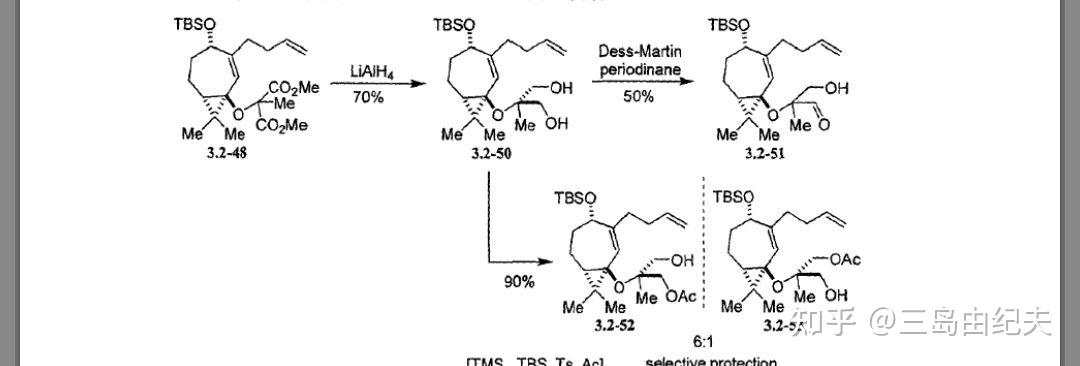
生成二叔基醚4.1-13后,与MeI在NaH条件下发生二酯α位的甲基化,再在LiAlH4条件下还原二酯生成二醇4.1-14,两步产率共72%。二醇4.1-14在Ac2O,Et2N条件下发生单甲酰化生成4.1-15-1,产率91%。4.1-15-1再用很经典的氧化醇为醛酮的Dess-Martin反应,氧化伯醇为醛基,得到4,1-16,产率95%。并且实现了由二酯的去对称化。

4.1-16在HF条件下发生分子内carbonyl-ene反应同时脱去TBS保护
醛酮做为亲烯体的Alder-ene反应被称为carbonyl-ene反应,生成高烯丙基醇化合物。Alder-ene反应机理类似于Diels-Alder反应,是周环反应的重要类型之一。连有吸电子基的亲烯体(比如顺丁烯二酸酐等),LUMO更容易参与反应,这和正常电子需求的Diels-Alder反应有相似之处。Alder-ene反应中烯体提供一个烯烃π键和烯丙基C-H σ键的四电子体系,亲烯体提供碳之间或碳与杂原子(比如C=O,C=N等),杂原子与杂原子(比如N=N,O=O等)之间的π键,经过六原子过渡态,六元环构象椅式最为稳定,也决定了反应的立体选择性,双键迁移最后形成新的C-H σ键和C-C σ键。在反应中在HF条件下4.1-16发生分子内carbonyl-ene反应构建了四氢呋喃环,并脱去TBS保护基得到(±)-Steenkrotin A 合成关键中间体4.1-17,产率90%。成功构建了[3,5,7]三环骨架,但是C7位羟基的构型与天然产物(±)-Steenkrotin A的构型不同,需要后续用氧化还原策略或Mitsunobu反应进行羟基构型的翻转。





至此已经完成了(±)-Steenkrotin A中关键的[3.4.5]三环骨架的构建,得到了关键合成中间体三环产物解决了极具挑战性的全取代的四氢呋喃环的合成。四环,五环骨架的构建在下一篇中介绍。


续:下一篇链接
